
The GRAS Process
REGULATORY IMPACT
In the early 1950s, Congress discovered that of the many hundreds of ingredients intentionally added to food, few had much scientific proof that the ingredients were safe. Congress faced the prospect of removal of these ingredients from the market until the Food and Drug Administration could review them and verify their safety.
FDA responded that review of the safety and testing data would take at least several months for each ingredient. Industry was up in arms at the Herculean cost in time and money for testing each ingredient. Congress faced a possible scenario where, until these ingredients were declared safe, FDA would be faced with an incredible log-jam of applications, industry would be overwhelmed financially, and consumers would be faced with very little variety on their plates.
Eventually, the questions became more focused: Why test all ingredients so rigorously, when some have been safely consumed for decades or, in the case of some natural products, hundreds or even thousands of years? Why must all ingredients, even those consumed in very small amounts, be tested to the same degree as ingredients consumed in large quantities? Why couldn’t some analogies be made with chemically similar ingredients known to be safe? And does it really require a government agency to determine when something is safe?
Congress responded with the 1958 Food Additives Amendment to the Federal Food, Drug, and Cosmetic Act. It divided food ingredients into two categories: those that are “generally recognized as safe” (GRAS) and those which are “food additives.” GRAS ingredients are those which have been found to be generally recognized as safe by “experts qualified by scientific training and experience to evaluate the safety of food and food ingredients. . . .” The ingredient must have had use in food prior to January 1, 1958, or be found safe by scientific procedures (i.e., testing data). Any food ingredient that could not be determined as GRAS by the above criteria would automatically be classified as a “food additive” and would require approval by FDA prior to addition to food—an often long and costly process.
If the ingredient was used prior to January 1, 1958, it could not have been as a drug or herbal remedy and must have been used by a significant number of people, to provide some assurance that at least most “less common” adverse reactions, such as allergies or metabolic intolerances, have been detected. Until the past two decades or so, the history of use also had to be within the U.S., but with increased exposure to other cultures and increased consumption of ethnic foods, the “historical use” requirements are becoming less restrictive.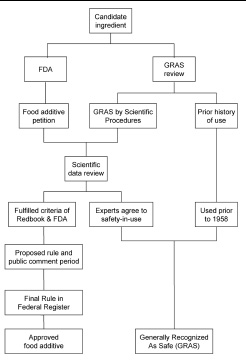
“Scientific procedures” usually means animal and/or human safety studies. Although the requirements may not be as stringent as those for a food additive petition, test data may include (1) the biochemical fate of a substance (e.g., metabolism, pharmacokinetics); (2) a repeated-dose study, such as a 90-day study in rats, (3) tests for reproductive effects, and (4) studies with bacteria to determine if the ingredient interacts with the DNA of the cells, often a sign of a cancer-causing ingredient. All of the animal tests are conducted at exaggerated doses to ensure that there is a margin of safety for the high-end human user, and the results are evaluated in terms of expected exposure to various demographic groups (e.g., children, the elderly). The success of a GRAS determination may hinge on an appropriate and accurate consumption analysis. The experts also examine the specifications of the ingredient, including the nature and amount of impurities present, the stability of the ingredient, and the method of manufacture or extraction from natural products.
The “experts qualified by scientific training and experience” may be convened by any source, including FDA, industry (any individual or corporation), or a trade association such as the Flavor and Extract Manufacturers Association (FEMA). GRAS recognition involves evaluation of the information by the experts, who reach a consensus as to whether a substance is GRAS. This GRAS determination is adequate to comply with the regulations and the law, but sometimes the manufacturer (or user) may take the additional step of notifying FDA of the GRAS finding. FDA may respond that it “has no questions” (i.e., does not object) or that the “notice does not provide a basis for a GRAS determination.” FDA notification is not a requirement but provides the manufacturer a layer of comfort that the agency is not in disagreement with the experts.
Of all the GRAS expert panels, the FEMA Expert Panel has had the greatest longevity (since 1960), and although it has confined its activities exclusively to flavor ingredients, it has recognized the most ingredients (nearly 2,000 substances) as GRAS (see “GRAS Flavoring Substances 21” on p. 46). As the FEMA example has shown for more than 40 years, the GRAS concept for evaluation of dietary ingredients is a model of industry and regulator cooperation.
by GEORGE A. BURDOCK
Principal, the Burdock Group
Vero Beach, Fla.