
Designing Clinical Trials to Substantiate Claims
Here’s what food scientists need to know about designing, conducting, or sponsoring clinical trials to substantiate health and structure/function claims.
Nutritional products have been welcomed by consumers, especially the health-oriented aging population—Baby Boomers—for many reasons, including higher awareness, increasing healthcare costs, ease of access, milder regulations, and relatively inexpensive prices. As a result, manufacturers and marketers of nutritional products are challenged with the necessity of conducting clinical trials to substantiate desired health claims or structure/function claims. Product developers wishing to make these claims for their products will have to either conduct clinical trials or sponsor clinical trials conducted for them under contract. This article explains the steps involved.

Types of Claims
In the United States, nutritional products, including Foods with Health Claims, Dietary Supplements, Foods for Special Dietary Uses, and Medical Foods are defined and regulated by the Food and Drug Administration (FDA). On the other hand, terms such as Nutraceuticals, Aquatic Nutraceuticals, Functional Foods, Functional Foods for Health, Designer Foods, Phytochemicals, Chemopreventive Agents, Antioxidants, and Enteral Nutrition are defined, not by FDA, but by various research, academic, industrial, and professional entities. These terms, although widely used in industry, do not have regulatory status yet.
In the international arena, the government of Japan has officially defined and regulates Foods for Special Health Use (FOSHU), and the government of Canada has defined the terms Functional Foods and Nutraceuticals. In Europe, the term VitaFoods is becoming popular, while the Codex Alimentarius Commission of the Food and Agriculture Organization(FAO) and the World Health Organization (WHO) of the United Nations has defined the term Foods for Special Medical Purposes.
Claims that can be used on food and dietary supplement labels fall into three categories: health claims, structure/function claims, and nutrient content claims. Only health claims and structure/function claims need to be substantiated by clinical trials (www.cfsan.fda.gov).
Health Claims
Health claims describe a relationship between a food, food component, or dietary supplement ingredient, and reduction in risk of a disease or health-related condition.
• Authorized Health Claims. The Nutrition Labeling and Education Act (NLEA) of 1990, the Dietary Supplement Act of 1992, and the Dietary Supplement Health and Education Act (DSHEA) of 1994 provide for health claims used on labels that characterize a relationship between a food, food component, dietary ingredient, or dietary supplement and risk of a disease, provided that the claims meet certain criteria and are authorized by an FDA regulation. FDA authorizes these types of health claims on the basis of an extensive review of the scientific literature, generally as a result of the submission of a health claim petition, using the Significant Scientific Agreement (SSA) standard to determine that the nutrient/disease relationship is well established (Table 1).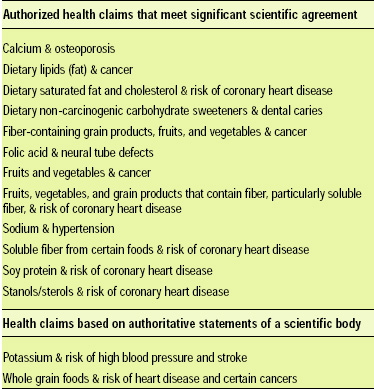
• Health Claims Based on Authoritative Statements. The Food and Drug Administration Modernization Act (FDAMA) of 1997 allows certain health claims to be made as a result of a successful notification to FDA of a health claim based on an “authoritative statement” from a scientific body of the U.S. Government or the National Academy of Sciences. FDAMA does not include dietary supplements in the provisions for health claims based on authoritative statements and therefore cannot be used for dietary supplements at this time (Table 1).
• Qualified Health Claims. FDA’s 2003 Consumer Health Information for Better Nutrition Initiative provides for the use of qualified health claims when there is emerging evidence for a relationship between a food, food component, or dietary supplement and reduced risk of a disease or health-related condition. In this case, the evidence is not well enough established to meet the SSA standard required for FDA to issue an authorizing regulation. Qualifying language is included as part of the claim to indicate that the evidence supporting the claim is limited. Both conventional foods and dietary supplements may use qualified health claims.
--- PAGE BREAK ---
FDA uses its enforcement discretion for qualified health claims after evaluating and ranking the quality and strength of the totality of the scientific evidence. Although FDA’s “enforcement discretion” letters are issued to the petitioner requesting the qualified health claim, the qualified claims are available for use on any food or dietary supplement product meeting the enforcement discretion conditions specified in the letter.
Table 2 lists the qualified health claims permitted by FDA, and Table 3 lists qualified health claim petitions received by FDA.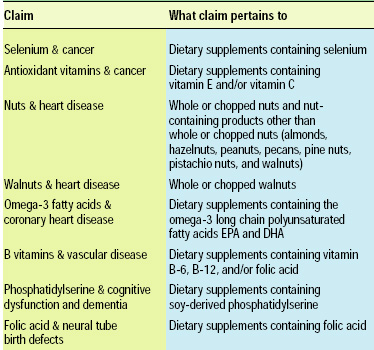

Structure/Function Claims
Structure/function claims describe the role of a nutrient or dietary ingredient intended to affect normal structure or function in humans, e.g., “Calcium builds strong bones.” In addition, they may characterize the means by which a nutrient or dietary ingredient acts to maintain such structure or function, for example, “Fiber maintains bowel regularity,” or “Antioxidants maintain cell integrity,” or they may describe general well-being resulting from consumption of a nutrient or dietary ingredient. Structure/function claims may also describe a benefit related to a nutrient-deficiency disease (like vitamin C and scurvy), as long as the statement also tells how widespread such a disease is in the U.S. The manufacturer is responsible for ensuring the accuracy and truthfulness of these claims; they are not pre-approved by FDA but must be truthful and not misleading.
• Dietary Supplements. If a dietary supplement label includes a structure/function claim, it must state in a “disclaimer” that FDA has not evaluated the claim. The disclaimer must also state that the dietary supplement is not intended to “diagnose, cure, mitigate, treat, or prevent any disease,” because only a drug can legally make such a claim. Manufacturers of dietary supplements that make structure/function claims on labels or in labeling must submit, no later than 30 days after marketing the dietary supplement, a notification to FDA that includes the text of the structure/function claim.
• Conventional Foods. Structure/function claims and disease claims for conventional foods focus on effects derived from nutritive value, while structure/function claims for dietary supplements may focus on nutritive as well as non-nutritive effects. Again, no drug claim should be made. FDA does not require conventional food manufacturers to notify FDA about their structure/function claims, and disclaimers are not required for conventional foods.
Clinical Trials
A clinical trial is a prospective study comparing the effect and value of an investigational product against a control (placebo) in human subjects. Clinical trials should be done by using the principles of Good Clinical Practice (GCP)—the international ethical and scientific quality standard for designing, conducting, recording, and reporting trials involving humans. Compliance with GCP provides public assurance that the rights, safety, and well-being of trial subjects are protected, and that clinical trial data are credible.
--- PAGE BREAK ---
GCP was developed by the International Conference on Harmonization of the technical requirements for registration of pharmaceuticals for human use, with consideration of the GCP of the European Union, Japan, U.S., Australia, Canada, Nordic countries, and WHO. In the U.S., this is published by FDA in a Guidance for Industry document and is used as the key reference. Fig. 1 is a schematic diagram of a clinical trial conducted according to GCP. The remainder of this article follows the path shown in the figure.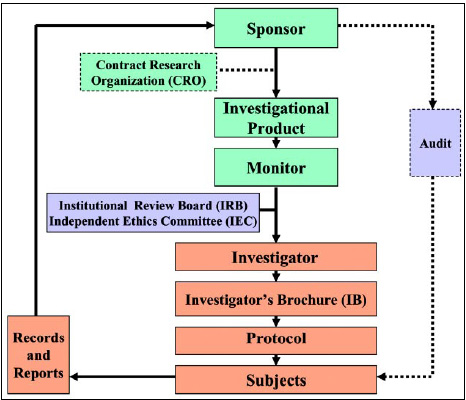
Prior to starting a clinical trial, manufacturers and marketers of nutritional products should:
• Compose the desirable claim for the product, and convert the claim into a hypothesis to be tested in a clinical trial.
• Conduct a comprehensive literature review and patent search to make sure that the testable hypothesis has not already been answered, and that the finding/solution has not been protected by patents. Collect all relevant information that becomes useful for the petition to FDA.
• Form a team of all collaborators to determine what tools, materials, personnel, time, budget, and subjects are necessary to test the hypothesis. Using the services of a Contract Research Organization (CRO) may be necessary at this stage.
• Devise methods to collect data from the trial subjects, and collect only the data needed.
• Consider conducting a pilot clinical trial to determine the feasibility of the main clinical trial. Pilot studies can be very helpful in revising parameters of a clinical trial.
Sponsor
The sponsor is an individual, company, or institution that takes responsibility for the initiation, management, and/or financing of a clinical trial.
• Responsibilities. The sponsor is responsible for implementing and maintaining Quality Assurance and Quality Control systems with written Standard Operating Procedures (SOPs) for all steps; ensuring that trials are conducted and data are generated, recorded, and reported in compliance with the protocol, GCP, and the applicable regulatory requirements; and securing agreement from all involved parties to ensure direct access to all trial-related sites, source data/documents, and reports, for the purpose of monitoring and auditing by the sponsor and inspection by regulatory authorities.
• Trial Management, Data Handling, and Recordkeeping. The sponsor should use qualified individuals to manage the overall conduct of the trial, handle and verify the data, conduct the statistical analyses, and prepare the trial reports. The sponsor should also ensure and document that electronic data processing systems conform to the sponsor’s established requirements for completeness, accuracy, reliability, and consistent intended performance (i.e., validation). The sponsor should use an unambiguous subject identification code that allows identification of all the data reported for each subject.
• Investigator Selection. The sponsor is responsible for selecting the investigator. Each investigator should be qualified by training and experience and should have adequate resources to properly conduct the trial. Prior to initiating a trial, the sponsor should define, establish, and allocate all trial-related duties and functions. Trial master files should be established at the beginning of the trial, at both the sponsor’s and investigator’s offices.
• Compensation to Investigators and Subjects. The financial aspects of the trial should be documented in an agreement between the sponsor and the investigator. If required by the applicable regulatory requirements, the sponsor should provide insurance or should indemnify (provide legal and financial coverage to) the investigator against claims arising from the trial, except for claims that arise from malpractice and/or negligence. The sponsor’s policies and procedures should address the costs of treatment of trial subjects in the event of trial-related injuries in accordance with the applicable regulatory requirements.
Contract Research Organization
The CRO is a person or organization (commercial, academic, or other) contracted by the sponsor to perform one or more of a sponsor’s trial-related duties and functions.
--- PAGE BREAK ---
When any or all of the duties and functions of the sponsor are transferred to a CRO, the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor. The CRO should implement quality assurance and quality control. Any trial-related duties and functions not specifically transferred to and assumed by a CRO are retained by the sponsor.
Investigational Product
An Investigational Product is an active ingredient or product (e.g., a nutritional product) or placebo being tested or used as a reference in a clinical trial.
When planning trials, the sponsor should ensure that sufficient safety and efficacy data from nonclinical and/or clinical trials are available to support human exposure at the dosages, for the duration, and in the trial population to be studied.
• Manufacturing, Packaging, Labeling, and Coding. The sponsor should ensure that the investigational product is manufactured in accordance with any applicable Good Manufacturing Practice (GMP) and is labeled and coded in a manner that protects the blinding (concealment of the investigational product’s identity), if applicable. The investigational product should be packaged to prevent contamination and an unacceptable level of deterioration during transport and storage. In blinded trials, the coding system for the investigational product should include a mechanism that permits rapid identification of the product in case of medical emergency, but does not permit undetectable breaks of the blinding.
• Supplying and Handling. The sponsor is responsible for supplying the investigator with the investigational product after the sponsor obtains all required documentation, such as approval/favorable opinion from the Institutional Review Board/Independent Ethics Committee (IRB/IEC). The sponsor should determine and inform all involved parties of acceptable storage conditions (e.g., temperature, relative humidity, and light) and expiration date. The sponsor should maintain records that document the shipment, receipt, disposition, return, and disposal of the investigational product.
• Safety Information. The sponsor is responsible for the ongoing safety evaluation of the investigational product. The sponsor should promptly notify the investigator and the regulatory authorities of findings that could adversely affect the safety of subjects, impact the conduct of the trial, or alter the IRB/IEC’s approval/favorable opinion to continue the trial.
Monitor
The monitor is an individual who oversees the progress of a clinical trial to verify that the rights and well-being of human subjects are protected, the reported trial data are accurate, complete, and verifiable from source documents, and the trial is conducted in compliance with the protocol and GCP.
• Selection. The monitor should be appointed by the sponsor, be appropriately trained, and have the scientific and/or clinical knowledge needed to monitor the trial adequately.
• Nature and Extent of Monitoring. The sponsor should determine the appropriate nature and extent of monitoring based on considerations such as the objective, design, complexity, blinding, size, and endpoints of the trial. In general, there is a need for on-site monitoring, before, during, and after the trial. Statistically controlled sampling may be an acceptable method of selecting the data to be verified.
• Responsibilities. The monitor should ensure that the trial is conducted and documented properly by carrying out activities, including acting as the main line of communication between the sponsor and the investigator; verifying that the investigator has adequate qualifications and resources, that staff and facilities, including laboratories and equipment, are adequate to safely and properly conduct the trial, and that these remain adequate throughout the trial period; verifying that the investigator follows the approved protocol; verifying that the investigator is enrolling only eligible subjects and that written informed consent was obtained before each subject’s participation in the trial; verifying that source data/documents and other trial records including Case Report Form are accurate, complete, and kept updated; and communicating to the investigator deviations from the protocol, SOPs, GCP, and the applicable regulatory requirements and taking appropriate action designed to prevent recurrence of the detected deviations.
• Monitoring Report. The monitor should submit a written report to the sponsor after each trial-site visit or trial-related communication. Reports should include the date, site, name of the monitor, and name of the investigator or other individuals contacted, and a summary of what the monitor reviewed and the monitor’s statements concerning the significant findings/facts, deviations and deficiencies, conclusions, actions taken or to be taken, and/or actions recommended to secure compliance.
--- PAGE BREAK ---
Audit
The purpose of a sponsor’s audit—which is independent of and separate from routine monitoring or quality control functions—should be to evaluate trial conduct and compliance with the protocol, SOPs, and GCP. The sponsor should appoint qualified auditors, who are independent of the clinical-trial/data-collection systems, to conduct audits.
• Auditing Procedures. The sponsor should ensure that auditing of the clinical trial is conducted in accordance with the sponsor’s written procedures on what to audit, how to audit, the frequency of audits, and the form and content of audit reports. The sponsor’s audit plan and procedures for a trial audit should be guided by the number of subjects in the trial, the type and complexity of the trial, the level of risks to the trial subjects, and any identified problems. The observations and findings of the auditor should be documented. Regulatory authorities may seek access to an audit report on a case-by-case basis, when evidence of serious GCP noncompliance exists.
• Noncompliance. Noncompliance with the protocol, SOPs, and GCP by an investigator, or by members of the sponsor’s staff, should lead to prompt action by the sponsor to secure compliance. If the monitoring and/or auditing identifies serious and/or persistent noncompliance on the part of an investigator, the sponsor should terminate the investigator’s participation in the trial and promptly notify the investigator, regulatory authorities, and the IRB/IEC.
Institutional Review Board/Independent Ethics Committee
The IRB is an independent body composed of medical, scientific, and non-scientific members that protects the rights, safety, and well-being of trial subjects by continuously reviewing all aspects of a clinical trial, including protocol.
The IEC is an independent body composed of medical, scientific, and non-scientific members which, by reviewing and providing favorable opinion on the trial protocol, investigator, facilities, and informed consent process, provides public assurance that the rights, safety, and well-being of trial subjects are protected.
• Composition. The IRB/IEC should consist of a reasonable number of members, who collectively have the qualifications and experience to review and evaluate the science, medical aspects, and ethics of the proposed trial. It is recommended that the IRB/IEC have at least five members, including at least one whose primary area of interest is in a non-scientific area and at least one who is independent of the institution/trial site. Only those members who are independent of the investigator and the sponsor of the trial should vote/provide opinion on a trial-related matter. A list of members and their qualifications should be maintained.
• Responsibilities and Functions. The IRB/IEC should safeguard the rights, safety, and well-being of all trial subjects; review the proposed clinical trial and document its views in writing (approval/favorable opinion; modifications required prior to its approval/favorable opinion; disapproval/negative opinion; or termination/suspension of any prior approval/favorable opinion); not admit any subject to a trial before it issues its written approval/favorable opinion of the trial; and retain all relevant records (e.g., written procedures, membership lists, lists of occupations/affiliations of members, submitted documents, minutes of meetings, and correspondence) for at least three years after completion of the trial and make them available on request from the regulatory authorities.
Investigator
The investigator is a person responsible for the conduct of the clinical trial.
• Qualifications and Responsibilities. The investigator should be qualified by education, training, and experience to assume responsibility for proper conduct of the trial; be thoroughly familiar with the appropriate use of the investigational product and comply with GCP and the applicable regulatory requirements, permit monitoring and auditing by the sponsor and inspection by the appropriate regulatory authorities; and have potential for recruiting the required number of suitable subjects within the agreed-on recruitment period, sufficient time to properly conduct and complete the trial within the agreed-on trial period, and adequate number of qualified staff and facilities to conduct the trial properly and safely for its foreseen duration.
• Medical Care of Trial Subjects. A qualified physician who is an investigator for the trial should be responsible for all trial-related medical decisions. During and following a subject’s participation in a trial, the investigator should ensure that adequate medical care is provided to a subject for any adverse events related to the trial. The investigator should also inform a subject when medical care is needed for intercurrent illness of which the investigator becomes aware. It is also recommended that the investigator inform the subject’s primary physician about the subject’s participation in the trial. Although a subject is not obliged to give his or her reason for withdrawing prematurely from a trial, the investigator should make a reasonable effort to ascertain the reason, while fully respecting the subject’s rights.
• Communication with IRB/IEC. Before initiating a trial, the investigator should have a written and dated approval/favorable opinion from IRB/IEC for the trial protocol, written informed consent form, and subject recruitment procedures (e.g., advertisements). During the trial, the investigator should provide to IRB/IEC all documents subject to its review.
--- PAGE BREAK ---
• Compliance with Protocol. The investigator should conduct the trial in compliance with the protocol agreed to by the sponsor and which was given approval/favorable opinion by IRB/IEC. The investigator and the sponsor should sign the protocol to confirm their agreement. The investigator should not implement any deviation from the protocol without agreement by the sponsor and prior review and documented approval/favorable opinion from IRB/IEC of an amendment (except where necessary to eliminate an immediate hazard to trial subjects). The investigators should maintain records that the subjects were provided the doses specified by the protocol, reconcile all investigational products received from the sponsor, and check, at appropriate intervals, that each subject is following the instructions properly.
• Randomization Procedures and Unblinding. The investigator should follow the trial’s randomization procedures and ensure that the code is broken only in accordance with the protocol. If the trial is blinded, the investigator should promptly document and explain to the sponsor any premature unblinding of the investigational product (e.g., accidental unblinding and unblinding due to a serious adverse event).
Investigator’s Brochure
The Investigator’s Brochure (IB) is a compilation of the nonclinical and clinical data on the investigational product that are relevant to the study of the product in human subjects. It is prepared by the sponsor to provide the investigator, IRB/IEC, and others involved in the trial with the information to facilitate their understanding of the rationale for, and their compliance with, many key features of the protocol, such as the dose, dose frequency/interval, and safety monitoring procedures. The information should be presented in a concise, simple, objective, balanced, and nonpromotional form that enables a clinician, or potential investigator, to understand it and make his or her own unbiased risk-benefit assessment of the appropriateness of the proposed trial.
The IB should provide a brief introductory statement highlighting the investigational product, bioactive ingredients, rationale for performing the research, physical and chemical properties, and formulation. It should provide the results of all relevant nonclinical and clinical studies, including safety (toxicological effects found in relevant studies), pharmacokinetics (findings on the absorption, distribution, metabolism and excretion of the investigational product), dose response (healthy volunteers and/or patients), efficacy, and adverse reactions for all the clinical trials.
Protocol
The protocol is a document that describes the objective, design, methodology, statistical considerations, and organization of a trial.
• General Information. The protocol should include the protocol title; identifying number; date; and name, title, address, telephone number, and e-mail address of the sponsor, monitor, sponsor’s medical expert for the trial, investigator, and the clinical laboratory and other medical and/or technical departments involved in the trial.
• Background Information. The protocol should also include the name and description of the investigational product, and a summary of findings from nonclinical studies that potentially have clinical significance, as well as from clinical trials that are relevant to the trial. It should also contain a summary of the known and potential risks and benefits to human subjects; a description of and justification for the dosage, dosage regimen, and treatment periods; the commitment to conduct the trial in compliance with the protocol and GCP; a description of the population to be studied, and references to literature and data that are relevant to the trial.
• Trial Objectives. A detailed description of the objectives of the trial is essential.
• Trial Designs and Procedures. The scientific integrity and credibility of the trial’s data depend substantially on the trial design. The Statement of End Points (primary and secondary) must be clear because it is the hypothesis and will be the item on which the merits of the trial are based.
Many different trial designs can be chosen, depending on the type of trial and the product being investigated. This article will discuss only three basic trial types:
Parallel design. Each group of subjects is expected to be given only one of the trial interventions (e.g., treatment or placebo). Parallel designs produce comparison between subjects.
Cross-over design. Each of the subjects is given the trial interventions in successive periods, usually after a washout period. Cross-over designs produce comparisons within subjects.
Factorial design. Two or more experimental interventions are evaluated not only separately but also in combination and against a placebo.
--- PAGE BREAK ---
The following points should be considered in all trial designs:
(1) A description of the measures taken to minimize/avoid bias:
Randomization: subjects are allocated at random to receive one of the interventions (treatment or placebo).
Blinding or masking: in a single-blind, only the investigator is aware of which intervention each subject is receiving; in a double-blind, neither the investigator nor the subject knows the identity of the intervention assignment.
Procedures for breaking codes.
(2) A description of the trial treatments; the dosage form, dosage regimen, packaging, and labeling of the investigational product; and the expected duration of subject participation.
(3) A description of the “stopping rules” or “discontinuation criteria.”
(4) Accountability procedures for the investigational products and placebo.
(5) Maintenance of trial treatment randomization codes and procedures for breaking codes.
(6) Identification of any data to be recorded directly on the Case Report Form and considered to be source data.
• Selection and Withdrawal of Subjects. The protocol must also consider inclusion criteria, exclusion criteria, withdrawal criteria, and dropouts.
• Assessment of Safety and Efficacy. Specification of safety and efficacy parameters and the methods and timing for assessing, recording, and analyzing safety parameters should be addressed.
• Statistics. A description of the statistical methods, sample size (number of subjects planned to be enrolled) and reason for choice of sample size, including reflections on (or calculations of) the power of the trial and level of significance should be provided. It is highly recommended that a biostatistician be involved as a member of the team designing the trial. Measurement of baseline data in subjects before the start of intervention; procedures for accounting for missing, unused, and spurious data; procedures for reporting any deviation from original statistical plan; and the selection of subjects to be included in the analyses should also be addressed.
• Direct Access to Source Data/Documents. The sponsor should ensure that the protocol specifies that the investigator will permit trial-related monitoring, audits, IRB/IEC review, and regulatory inspection by providing direct access to source data/documents.
• Other Considerations. Ethical considerations, insurance coverage, and publication policy (papers and patents) must be addressed in the protocol, if not addressed in a separate agreement.
Subjects
A subject (participant or partner) is an individual who participates in a clinical trial, either as a recipient of the investigational product or as a control.
• Recruitment of Trial Subjects. The investigator should have IRB/IEC’s written approval/favorable opinion of the written informed consent form to provide to subjects.
--- PAGE BREAK ---
Neither the investigator, nor the trial staff, should coerce or unduly influence a subject to participate, or continue to participate, in a trial.
The written informed consent form should not contain any language (including foreign words) that causes the subject to waive any legal rights, or that releases the investigator, the sponsor, or their agents from liability for negligence.
The language used in the consent form should be as nontechnical as practical and should be understandable to the subject.
Before informed consent may be obtained, the investigator should provide the subject ample time and opportunity to inquire about details of the trial and to decide whether or not to participate in the trial. All questions about the trial should be answered to the satisfaction of the subject.
• Written Informed Consent Form. Prior to a subject’s participation in the trial, the consent form should be signed and personally dated by the subject and by the person who conducted the informed consent discussion.
The consent form should clearly explain that the trial involves research and that the subject’s participation is voluntary. It should also explain the purpose of the trial, the trial treatments, the probability for random assignment to each treatment, the trial procedures to be followed, the subject’s responsibilities, the reasonably foreseeable risks or inconveniences to the subject, the potential benefits to the subject, the alternative procedures or courses of treatment that may be available to the subject, and their important potential benefits and risks.
The consent form should specify the compensation and/or treatment available to the subject in the event of trial-related injury, the anticipated prorated payment to the subject for participating in the trial, and the anticipated expenses to the subject for participating in the trial and state that the subject may refuse to participate or withdraw from the trial at any time without penalty or loss of accrued benefits.
Prior to participation in the trial, the subject should receive a copy of the signed and dated consent form. If the results of the trial are published, the subject’s identity will remain confidential.
Records and Reports
The investigator should ensure the accuracy, completeness, legibility, and timeliness of the data recorded in Case Report Form (CRF) and in all required reports. Data reported in the CRF, which are derived from source documents, should be consistent with the source documents, or the discrepancies should be explained. Any change or correction to a CRF should be dated, initialed, and explained, and should not obscure the original entry (i.e., an audit trail should be maintained). This applies to both written and electronic changes and corrections.
The investigator should maintain the trial documents and take measures to prevent their accidental or premature destruction. Essential documents should be retained for at least three years after the formal discontinuation of clinical development of the investigational product. It is the responsibility of the sponsor to inform the investigator as to when these documents no longer need to be retained.
On request of the sponsor, monitor, auditor, IRB/IEC, or regulatory authority, the investigator should make available for direct access all requested trial-related records. Also, the financial aspects of the trial should be documented in an agreement between the sponsor and the investigator.
The investigator should submit progress reports to the IRB/IEC at least annually. The investigator should also promptly provide written reports to the sponsor and IRB/IEC on any changes significantly affecting the conduct of the trial and/or increasing the risk to subjects.
--- PAGE BREAK ---
• Safety Reporting. An Adverse Event (AE) is any untoward medical occurrence in a subject who is being administered an investigational product that does not necessarily have a causal relationship with this treatment. An AE can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of an investigational product, whether or not related to the investigational product. From the time of consent until the end of the trial, every AE must be recorded and reported to the sponsor immediately.
A Serious Adverse Event (SAE) is any untoward medical occurrence that at any dose results in death, is life-threatening, requires inpatient hospitalization or prolongation of existing hospitalization, results in persistent or significant disability/incapacity, or is a congenital anomaly/birth defect. All SAEs should be reported immediately to the sponsor, IRB/IEC, and the regulatory authorities.
• Premature Termination or Suspension of a Trial. If the trial is terminated prematurely or suspended for any reason, the investigator should promptly inform the trial subjects, assure appropriate therapy and followup for the subjects, and inform the sponsor, IRB/IEC, and the regulatory authorities.
• Final Report by Investigator. On completion of the trial, the investigator should provide the sponsor with a detailed final report, including an explanation of the trial’s outcome; a summary should be provided to IRB/IEC. The investigator may also suggest application for patents, if warranted, and publication of the results in scientific journals.
Petition to FDA
The sponsor should include the investigator’s final report as evidence in support of a petition to FDA for substantiating desirable health claims. The sponsor may also use the investigator’s final report in support of marketing efforts for structure/function claims. FDA encourages petitions and notifications to be submitted electronically.
For additional reading on this topic, see the online version of this issue of Food Technology at www.ift.org.
by A. Reza Kamarei and Carl Trygstad
Author Kamarei, a Professional Member of IFT, is Interim Vice President of R&D and Director of Nutrition, Oceanic Institute, 41-202 Kalanianaole Hwy., Waimanalo, HI 96795 ([email protected]). Author Trygstad is Vice President of Medical and Scientific Services, Quintiles Transnational Japan-Hawaii, 745 Fort St., Honolulu, HI 96813 ([email protected]). Send reprint requests to author Kamarei.