
Bioprocess Engineering of Enzymes
The increasing availability of new bioengineering techniques makes for a very promising future for the development of new enzymes to meet the demands of the food industry.
The term enzyme (which means “in leaven”) was not part of our scientific vocabulary until 1878, when it was coined by Kuhne. The effects of enzymes as agents of chemical, physical, and functional change in foods, however, had been observed and exploited for several centuries earlier in products like cheese, meat, beer, wine, baked goods, etc.
For example, Greek epic poems dating back to 800 B.C. had made reference to the role of enzymes in cheese production, and Spallanzani observed meat digestion by hawk gastric juice in 1783. The ancient practice by South and Central American natives of tenderizing meat by wrapping them in papaya leaves, and production of Chinese rice wines and Japanese sake using crude preparations of what are now known as papain and amylase, respectively, are further examples of the use of enzymes before the advent of modern science unraveled the “mystery” of these practices.
The broad range of applications of enzymes in the food industry coupled with the advantages of enzyme technology (nature’s own resource, specificity, and mild operating conditions) over other, nonenzymatic processes provided the impetus for commercial enzyme production. Initial products were based on the use of plants and animals as primary sources of raw material, but competition from other, more lucrative applications for these resources, coupled with product inconsistencies resulting from seasonal, climatic, and other agricultural variables, made the cost of production prohibitive.
These problems paved the way for exploiting microorganisms as a major source for enzymes because of their rapid multiplication and thus high enzyme yields over short periods. In 1891, Japanese biochemist Jokichi Takamine developed and patented the first surface-culture fermentation process for industrial production of fungal amylase. Commercial enzyme production was given a tremendous boost with the development of submerged fermentation in the mid-20th century—particularly production of amyloglucosidase in the 1960s—and it has become the method of choice for most commercial enzyme products derived from microbial sources (see Table 1).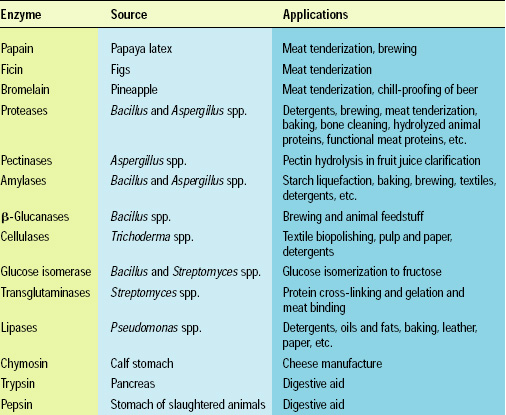
Industrial Enzyme Production
The primary step in enzyme production involves selection of enzyme source by screening plants, animals, and microbes (bacteria, archaea, fungi) from all ecological niches, ranging from “normal environments” to extreme habitats like volcanoes, cold Arctic regions, hydrothermal vents in the deep sea, soda and saline lakes, etc. The choice of a particular environment for any enzyme is largely determined by the conditions of the projected industrial application. For example, soda lakes and hot spring environments will be more likely sources for microorganisms (alkalophiles) expressing enzymes capable of working under alkaline and high-temperature conditions, while those from the cold Arctic environment (psychrophiles) are more suitable for cold-temperature applications, having been adapted to such conditions in the evolutionary process (Duckworth et al., 1996). Samples from selected environments are subsequently subjected to primary and secondary screenings based on sensitive and selective enzyme assays, followed by purification and characterization of the best candidates.
The microorganisms used may be divided into two main categories: wild-type organisms, which are the original sources of the enzyme of interest and produce “wild-type” enzymes, and recombinant expression systems or genetically modified organisms (GMOs), in which the gene for the enzyme is transferred from its original source and cloned in the genome of another organism for optimal expression; thus, while the expression organism is genetically modified, the expressed enzyme is not.
The vast majority of microorganisms thus far exploited for industrial enzyme production belong to a rather small cluster of so-called taxonomic “hot spots.” These are Bacillus and Pseudomonas species from the bacterial kingdom and Aspergillus, Fusarium, Trichoderma, and Mucor from the fungal kingdom (Dalboge and Lange, 1998). Generally, these microorganisms must meet regulatory standards and be safe strains, which implies nontoxic, nonpathogenic, and non-antibiotic- producing, for the enzyme products to be permitted for food and other applications.
--- PAGE BREAK ---
Traditionally, the source organism expressing the activity of interest is used to inoculate the culture medium and after scale-ups is transferred into a production fermenter under controlled conditions of pH, temperature, agitation, air flow rate, nutrient quantity, etc. (Fig. 1). The fermentation is constantly monitored via automated systems until the desired conditions signifying completion of the process are realized. The fermentation broth, then made up of unused nutrients, microorganisms, water, and enzymes, is subjected to downstream processing for product recovery.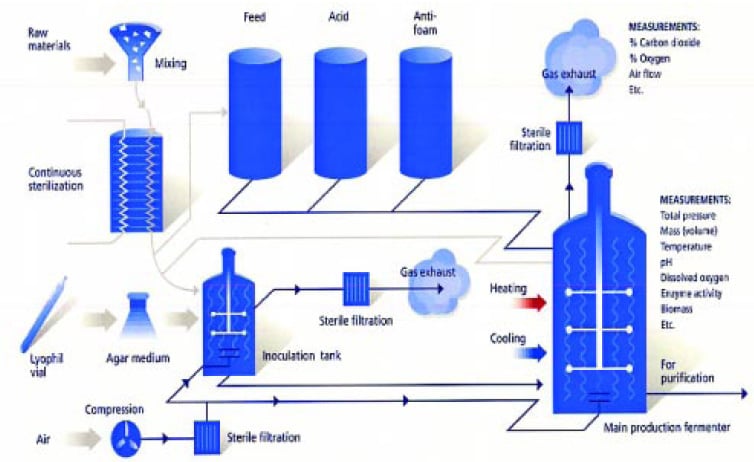
The recovery strategy is largely dependent on the projected application and is more stringent for diagnostic and therapeutic enzymes than for bulk industrial enzymes like lipases, cellulases, amylases, and proteases. The techniques used include centrifugation, homogenization (for cell disruption in the case of intracellular enzymes), filtration, and ultrafiltration to concentrate the cell-free extract. Stabilizers are also added to the extract and stored in liquid form or packaged in solid form by spray drying, drum drying, or encapsulation. Ideally, the use of the original source organism for expressing an enzyme should provide the best scenario for enzyme production because, in principle, it would speed up the process and reduce development cost. However, it has some limitations: the original organisms often express other side activities which may limit the range of applications, and fermentation yields are often too low.
Role of Recombinant DNA Technology
The advent of recombinant DNA technology in the 1980s greatly revolutionized commercial enzyme production, making it possible to address some of the problems in using recombinant microorganisms. At the core of recombinant technology is gene cloning, which involves cutting DNA from an organism with a desired gene into linear fragments and inserting it into a plasmid vector, usually with a high frequency of replication (high copy number). The resulting recombinant plasmids are used to transform Escherichia coli or some other microorganism to create a DNA library of recombinant clones, from which those containing the desired gene may be selected.
Once the best candidate expressing the desired enzyme activity is selected, the gene is sequenced and ultimately transferred into production strains, such as Bacilli for genes of prokaryotic origin or fungal species such as Aspergillus or Trichoderma for eukaryotic genes (Fig. 2). In the current state of the art, these production strains are highly optimized, having been subjected to classical strain improvement by mutagenesis using chemicals or irradiation. These strains and expression cassettes used enable the industry to express desired enzymes in large quantities with minimal or completely eliminated side activities.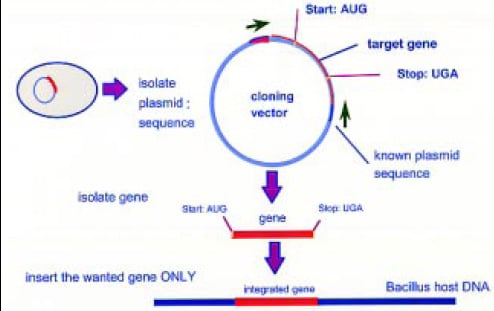
Initial attempts to clone eukaryotic genes (e.g., fungi) in prokaryotes were, however, unsuccessful because of the presence of introns (noncoding sequences) in eukaryotic genomes which had to be edited out before transcription. The inability of prokaryotes to recognize and edit out the introns meant that the translated protein lacked the expected characteristics. This was addressed by the technique of cDNA cloning, in which the transcribed mRNA devoid of introns is isolated instead of DNA (Johnson-Green, 2002). The use of cDNA cloning paved the way for expressing chymosin (from bovine source) in Aspergillus nidulans and Mucor miehei. Several other enzymes have since been produced using this technology.
A modification of cDNA cloning referred to as expression cloning was later developed to speed up the isolation and characterization of genes from filamentous fungi (Dalboge and Heldt-Hansen, 1994). This method (Fig. 3 on p. 48) combines the ability of Saccharomyces cerevisiae to express heterologous genes with sensitive, reliable, and simple enzyme assays. Hundreds of fungal genes expressing wide-ranging activities, including xylanases, proteases, galactanases, lipases, mannanases, and pectin-degrading enzymes, have been identified and cloned in Aspergillus oryzae using this method (Dalboge and Lange, 1998).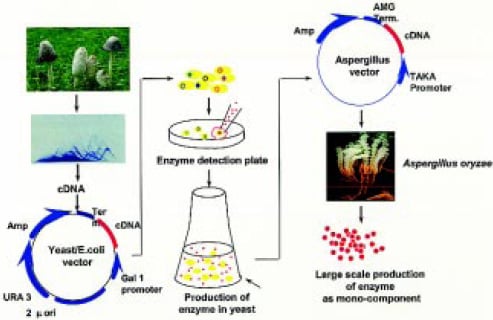
--- PAGE BREAK ---
Expression cloning has the following advantages over standard cDNA cloning: it is independent of any prior knowledge of the enzyme to be cloned, thus eliminating the purification and characterization steps required prior to cloning, and it allows for simultaneous screening for several different enzymes by simply changing the substrate in the agar plates.
While the techniques discussed above have ensured the production of a wide range of enzymes for food and other industrial applications, new developments in the field of molecular biology have opened up further opportunities which speed up the process of enzyme discovery and allow the development of engineered or hybrid enzymes. These “customized” enzymes tend to have improved properties, such as pH stability, thermostability, increased activity, and enhanced interaction with substrates, which address specific demands of the food and other industries. The novel techniques, some of which are described below, may be divided into two main categories: screening methods (for natural diversity), and mutagenesis or artificial evolution.
Screening Methods
Among the screening methods are molecular screening and bioinformatics.
• Molecular Screening. This method identifies enzyme genes independently of enzyme expression levels by using degenerate primers to identify conserved regions from the genomic DNA. Sequence information from a known gene expressing an activity of interest is used to screen for homologous genes from other sources. The identified homologous regions are used to design polymerase chain reaction (PCR) primers to amplify and sequence the newly identified gene. Thus, by growing many different organisms and isolating and analyzing their DNA using the PCR primers, several genes encoding the desired activity may be identified without the need to assay for enzyme activity. The method has been used to identify Fam45 fungal genes encoding cellulases from several ecologically and taxonomically diverse species (Henrissat and Romeau, 1995; Geremia et al., 1996).
• Bioinformatics. While the traditional process of searching for new enzymes has been based on specimen collection and laboratory experimentation, bioinformatics identifies such enzymes through data mining (i.e., data retrieval and integration) of proprietary and publicly available databases. The stored information in these databases (e.g., LIGAND, BRENDA, TIGR) covers a range of fields, including functional genomics and proteomics. Genomics deals with DNA/genome sequences and derivation of information from such sequences by computer analysis, while functional genomics defines the status of the relationship between the transcribed mRNA profiles and the corresponding proteome (or translated protein). Proteomics deals with identification and quantification of proteins, their sequences, localization, post-translational modifications, interactions, structure, and function (Bull et al., 2000).
Data mining for information from these databases is accomplished in one of two ways: explicit searching and “shotgun” searching. The former is very precise and provides well-defined search parameters which filter out noise in the data while highlighting the characteristics of interest. For example, a search specifying a gene or protease expression based on a specific assay, ligand-protease/DNA interaction, or electrophoresis maps, generates very finely filtered data; this significantly reduces the target set of genes to be explored experimentally and consequently speeds up the screening process. However, most searches often begin with the “shotgun” approach, in which several databases are searched on a broad range of criteria and the information obtained is filtered a couple of times to eliminate noise (Bull et al., 2000).
Bioinformatics in combination with other screening methods has proved to be a very powerful tool for enzyme discovery. For example, it has been used to compile a list of the approximately 4,100 genes in Bacillus subtilis.
Mutagenesis
Enzymes do not always function optimally under the application conditions because of such factors as thermostability, pH stability, and compatibility to chemicals and other ingredients in the process. Such limitations may be addressed by rescreening of bacteria and/or fungi for a better enzyme or improving enzyme characteristics by gene manipulation. The principal methods currently used for gene manipulation are rational protein engineering and random mutagenesis, which is also referred to as directed or artificial evolution because it mimics the natural evolutionary process. It is important to note that the two methods, while discussed separately here, are complementary to or even supportive of each other and therefore often combined in their applications (Kirk et al., 2002).
--- PAGE BREAK ---
Site-directed mutagenesis has been used since the 1980s to engineer new enzymes with modified characteristics. This utilizes protein-engineering and computer-modeling techniques to predict the impact of specific amino acids on the three-dimensional structure of an enzyme, its pH and temperature optimum/stability profiles, and substrate specificity. Site-directed mutagenesis of the corresponding DNA with pre-designed oligonucleotide primers results in expression of enzymes with the desired characteristics (Garvey and Matthews, 1990; Wagner and Benkovic, 1990). Table 2 shows some examples of enzyme modifications using this method. The accuracy and rate of such computer-generated predictions of enzyme structure may be limited by the limited knowledge it provides on protein structure–function relationships; the imprecise nature of computer modeling; and the large numbers of mutants that need to be screened.
Several artificial evolution techniques developed within the past decade have greatly enhanced the process of gene manipulation. These are discussed below. Other available techniques that the reader may refer to are sequence homology-independent protein recombination (SHIPREC), a combination of ITCHY and DNA shuffling into a single method referred to as SCRATCHY, random chimeragenesis on transient templates (RACHITT), and staggered extension process (StEP) (Pelletier, 2001; Lutz et al., 2001; Sieber et al., 2001; Zhao et al., 1998).
• Error-Prone PCR.This takes advantage of inherent inaccurate copying by DNA polymerases and the reduced fidelity of DNA polymerase by manganese ions to introduce point mutations into genes during PCR (Lin-Goerke et al., 1997). The procedure relies on inclusion of Mn ions instead of Mg, reduced concentration of nucleotides, and increased number of PCR cycles to increase the probability of nucleotide mis-insertion to ensure point mutations. A wide range of substitutions can be obtained by this process, including AT to GC and GC to AT transitions, as well as AT to TA transversions (Fromant et al., 1995). Because of the changes in error-prone PCR being intragenomic, it is also referred to as “asexual PCR.” Mutagenized enzymes are screened for desired activity and/or characteristics, cloned, and eventually expressed. Error-prone PCR is, however, rather slow, introducing only single-point mutations, and is limited by the small size of fragments (less than 800 base pairs) amenable to the process.
• DNA Shuffling. Unlike error prone PCR, DNA shuffling is described as “sexual PCR,” since it allows mixing of genetic material between different parent sequences (Fig. 4). Parent sequences with desired traits are broken down into random fragments of double- stranded DNA, and the fragments are subjected to repeated cycles of denaturation, annealing, and extensions in the presence of DNA polymerase to obtain full-length chimeras. Subjecting these to regular PCR amplifies the shuffled strands, from which a library may be generated and screened (Stemmer, 1994a, b; Moore and Maranas, 2000). Table 2 shows some examples where this technique has been used to modify enzyme characteristics.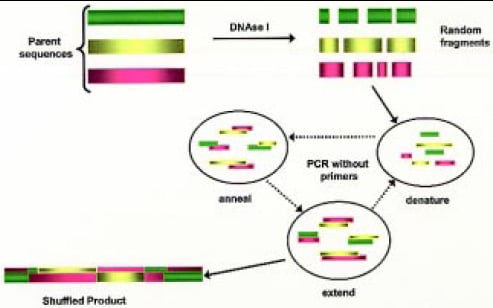
A modification to DNA shuffling referred to as exon shuffling provides another tool for rapid evolution of enzymes and other biomolecules. This takes advantage of the presence of coding (exons) and noncoding (introns) sequences in eukaryotic genomes and the encoding of separate folding domains per exon. Thus, recombination by inserting a desired gene within introns results in assembly of independent exons into a chimeric or hybrid single gene, which is consequently expressed as a novel enzyme with unique characteristics (Patthy, 1999; Nixon et al., 1997). This has been used to develop and express a single hybrid enzyme from exons of a hydrolase and glycinamide ribonucleotide transformylase. The resulting specific activity of the hybrid was 100–1,000 times that of the wild-type transformylase (Nixon et al., 1997).
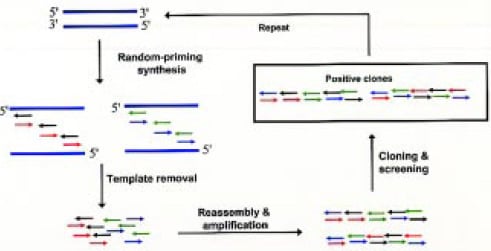
• Random-Priming Recombination (RPR). This technique (Fig. 5) uses random sequence primers to generate large numbers of short DNA fragments complementary to different segments of the template sequence. These DNA fragments may contain low levels of point mutations as a result of base mispriming or mis-incorporation. Following removal of the template, the short fragments can prime one another based on sequence homology and be recombined into full-length genes by repeated thermocycling in the presence of DNA polymerase. The resulting-full length genes may be further amplified by PCR and cloned into a vector for expression, followed by screening and selection of recombinants with desired activity (Shao et al., 1998).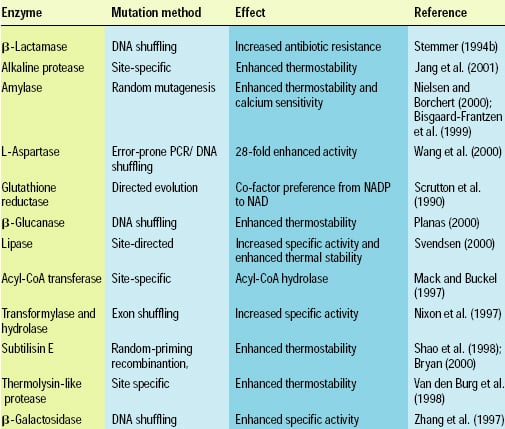
--- PAGE BREAK ---
• Incremental Truncation (ITCHY).This is a combinatorial approach to creating hybrid enzyme libraries by allowing shuffling of genes independent of sequence homology (Ostermeier et al., 1999), and it has the advantage of constructing a library containing all possible truncations of a gene, gene fragment, or DNA library in a single experiment (Fig. 6).
The process involves protecting one end of a linear DNA containing the gene of interest, while the other end is made susceptible to Exonuclease III digestion. The digestion is made to proceed at a very slow rate, which allows aliquots taken at frequent intervals to have a DNA library of all possible single codon or base pair deletions. The ends of the truncated DNA library are blunted, ligated, and ultimately expressed.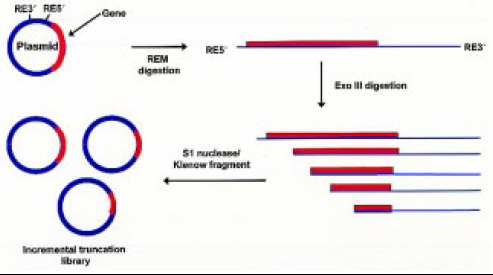
The method provides solutions to two key issues in directed evolution of enzymes or biomolecules: where to fuse enzymes or enzyme fragments to produce active hybrids, since it is able to identify functional structural motifs such as binding and catalytic domains; and what are the bisection points of enzymes to ensure expression of desired characteristics (Ostermeier et al., 1999).
• Random Insertion and Deletion (RID). This method randomly introduces codon-based mutations along the entire range of a target gene by base deletions, while simultaneously inserting other gene fragments or entire genes in the same position (Murakami et al., 2002). It involves fragmentation of the parent gene with restriction enzymes, followed by ligation of the fragment to a linker. The product is then digested to make a linear dsDNA with a break in the antisense chain. This dsDNA is cyclized using DNA ligase, and the circular dsDNA is treated with DNA polymerase to produce a circular ssDNA, which is randomly cleaved at single positions with a Ce(IV)-EDTA complex.
The resulting linear ssDNAs are ligated with 5' and 3' anchors at both ends, and the anchored DNAs are amplified by PCR. These are digested by restriction enzymes to introduce base deletions, followed by fragmentation and cyclization in presence of DNA polymerase (Klenow fragment) and ligase, respectively. The genes are then cloned and screened for expression of desired characteristics. RID has been used to produce mutants of a green fluorescent protein that could not be generated by error-prone PCR (Murakami et al., 2002).
Future Prospects
Enzymes have evolved through the years to become an integral part of many food processing operations. This is due, in large part, to the desire for developing more “environmentally friendly” solutions to the limitations of some of these operations. Bioprocess engineering of enzymes using the highly innovative and rapidly expanding toolbox in recombinant DNA technology has been at the core of most of these developments and has provided the food industry with “customized enzyme” solutions to address specific problems, some of which are listed in Table 2.
In spite of the successes made using these tools, it is still a rapidly evolving field, having broken ground only in the past two decades. The increasing availability of new techniques and equipment for high-throughput screening and analytical operations and the increasing and readily accessible genomics and proteomics information certainly make for a very promising future for the development of new enzymes to meet the demands of the food industry.
by Isaac N.A. Ashie
The author, a Professional Member of IFT, is Senior Scientist, Application Technology, Novozymes North America Inc., 77 Perry Chapel Church Rd., Franklinton, NC 27525.
References
Bisgaard-Frantzen, H., Svendsen, A., Norman, B., Pedersen, S., Kjaerulff, S., Outtrup, H., and Borchert, T.V. 1999. Development of industrially important α-amylases. J. Appl. Glycoscience 46: 199-206.
Bryan, P.N. 2000. Protein engineering of subtilisin. Biochimica et Biophysica Acta 1543: 203-222.
Bull, A.T., Ward, A.C., and Goodfellow, M. 2000. Search and discovery strategies for biotechnology: The paradigm shift. Microbiol. Mol. Biol. Rev. 64: 573-606.
Dalboge, H. and Lange, L. 1998. Using molecular techniques to identify new microbial biocatalysts. TIBTECH 16: 265-272.
Dalboge, H., and Heldt-Hansen, H. 1994. A novel method for efficient expression cloning of fungal enzyme genes. Mol. Gen. Genet. 243: 253-260.
Duckworth, W.W., Grant, W.D., Jones, B.E., and van Staanbergen, R. 1996. Phylogenetic diversity of soda lake alkaliphiles. FEMS Microbiol. Ecol. 19: 181-191.
Fromant, M., Blanquet, S., and Plateau, P. 1995. Direct random mutagenesis of genesized DNA fragments using polymerase chain reaction. Anal. Biochem. 224: 347-353.
Garvey, E.P. and Matthews, C.R. 1990. Site-directed mutagenesis and its application to protein folding. Biotechnology 14: 37-63.
Geremia, R.A., Petroni, E.A., Ielpi, L., and Henrissat, B. 1996. Towards a classification of glycosyltransferases based on amino acid sequence similarities: prokaryotic amannosyltransferases. Biochem. J. 318: 133-138.
Henrissat, B. and Romeau, A. 1995. Families, superfamilies and subfamilies of glycosylhydrolases. Biochem. J. 311: 350-351.
Jang, J.W., Ko, J.H., Kim, E.K., Jang, W.H., Kang, J.H., and Yoo, O.J. 2001. Enhanced thermal stability of an alkaline protease, AprP, isolated from a Pseudomonas sp. by mutation at an autoproteolysis site, Ser-331. Biotechnol. Appl. Biochem. 34: 81-84.
Johnson-Green, P. 2002. Gene cloning and production of recombinant proteins. In “Introduction to Food Biotechnology,” ed. F.M. Clydesdale, pp. 45-79. CRC Press, Boca Raton, Fla.
Kirk, O., Borchert, T.V., and Fuglsang, C.C. 2002. Industrial enzyme applications. Curr. Opin. Biotech. 13: 1-7.
Lin-Goerke, J.L., Robbins, D.J., and Burczak, J.D. 1997. PCR-based random mutagenesis using manganese and reduced dNTP concentration. Biotechniques 23: 409-412.
Lutz, S., Ostermeier, M., Moore, G.L., Maranas, C.D., and Benkovic, S.J. 2001. Creating multiple-crossover DNA libraries independent of sequence identity. Proc. Natl. Acad. Sci. USA. 98: 11248-11253.
Mack, M. and Buckel. W. 1997. Conversion of glutaconate CoA-transferase from Acidaminococcus fermentans into an acyl-CoA hydrolase by site-directed mutagenesis. FEBS Lett. 405: 209-212.
Moore, G.L. and Maranas, C.D. 2000. Modeling DNA mutation and recombination for directed evolution experiments. J. Theor. Biol. 205: 483-503.
Murakami, H., Hohsaka, T., and Sisido, M. 2002. Random insertion and deletion of arbitrary number of bases for codon-based random mutation of DNAs. Nature Biotech. 20: 76-81.
Nielsen, J.E. and Borchert, T.V. 2000. Protein engineering of bacterial a-amylases. Biochimica Biophysica Acta 1543: 253-274.
Nixon, A.E., Warren, M.S., and Benkovic, S.J. 1997. Assembly of an active enzyme by the linkage of two protein modules. Proc. Natl. Acad. Sci. USA. 94: 1069-1073.
Ostermeier, M., Nixon, A.E., and Benkovic, S.J. 1999. Incremental truncation as a strategy in the engineering of novel biocatalysts. Bioorganic Med. Chem. 7: 2139-2144.
Patthy, L. 1999. Genome evolution and the evolution of exon shuffling—A review. Gene 238: 103-114.
Pelletier, J.N. 2001. A RACHITT for our toolbox. Nature Biotechnol. 19: 314-315.
Planas, A. 2000. Bacterial 1,3-1,4-β-glucanases: Structure, function and protein engineering. Biochimica Biophysica Acta. 1543: 361-382.
Scrutton, N.S., Berry, A., and Perham, R.N. 1990. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343: 38-43
Shao, Z., Zhao, H., Giver, L., and Arnold, F.H. 1998. Random-priming in vitro recombination: An effective tool for directed evolution. Nucl. Acids Res. 26: 681-683.
Sieber, V., Martinez, C.A., and Arnold, F.H. 2001. Libraries of hybrid proteins from distantly related sequences. Nature Biotechnol. 19: 456-460.
Stemmer, W.P.C. 1994a. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. USA. 91:10747-10751.
Stemmer, WPC. 1994b. Rapid evolution of a protein in vitro by DNA shuffling. Nature 370: 389-391.
Svendsen, A. 2000. Lipase protein engineering. Biochimica et Biophysica Acta. 1543: 223-238.
Van den Burg, B., Vriend, G., Veltman, O.R., Venema, G., and Eijsink, V.G.H. 1998. Engineering an enzyme to resist boiling. Proc. Natl. Acad. Sci. USA. 95: 2056-2060.
Wagner, C.R. and Benkovic, S.J. 1990. Site-directed mutagenesis: A tool for enzyme mechanism dissection. Trends Biotechnol. 8: 263-270.
Wang, L-J, Kong, X-D, Zhang, H-Y, Wang, X-P, and Zhang, J. 2000. Enhancement of the activity of L-aspartase from Escherichia coli W by directed evolution. Biochem. Biophys. Res. Commun. 276: 346-349.
Zhang, J.H., Dawes, G., and Stemmer, W.P.C. 1997. Evolution of an effective fucosidase from a galactosidase by DNA shuffling. Proc. Natl. Acad. Sci. USA. 94:4504-4509.
Zhao, H., Giver, L., Shao, Z., Affholter, J.A., and Arnold, F.H. 1998. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nature Biotechnol. 16: 258-261.