Is It a Food, Dietary Supplement, or Drug?

Many foods have drug-like properties. For example, foods with vitamin C (such as citrus fruit) can cure scurvy, the dreaded disease of 17th century sailors; likewise, seafood can cure (thyroid) goiters because seafood represents a source of iodine, almost unique in nature. Most obviously, food can cure malnutrition and starvation, and protein-containing foods are a remedy for kwashiorkor (a protein deficiency disease). Cranberries contain anthocyanins, which help maintain a healthy urinary tract, essentially through the creation of an unfavorable environment for urinary tract pathogens. Fermentable fiber found in peas, beans, and bran, once acted upon by gut microflora creates an unfavorable colonic environment for pathogens such as Clostridium difficile, a notable killer in hospices. Omega-3 fatty acids from marine plants and fish are important in preventing cardiovascular disease. Raw cotton seed oil has antimalarial properties and contains gossypol, which can act as a male contraceptive, but refined cotton seed oil was used to make Crisco shortening. Theophylline, which is found in chocolate, is toxic to dogs but is used as a bronchodilator for asthmatics. Indeed, many foods have drug-like properties.
Figure 1. Possible Regulatory Categories. From Burdock Group
And as consumers seek out more food and beverage products with high nutrition, food marketers are answering the demand by creating products with health-giving properties. But are these products new generation healthful foods, dietary supplements, or drugs?
A common question among ingredient and supplement manufacturers who seek advice from Burdock Group, a safety and regulatory consulting firm, is this: If their newly isolated extract of an edible plant is still a food, can it be used as a supplement, even though it has drug-like properties? And given the fact that the substance occurs naturally and because it has been consumed for centuries, is it therefore exempt from classification as a drug (Figure 1)?

Figure 2. Regulatory Framework Drivers. From Burdock Group.
A major determinant of the classification is how the manufacturer characterizes the product on its label, (i.e., its intended use), which drives the decision by the U.S. Food and Drug Administration (FDA) on how the substance is regulated. This article will describe FDA’s regulatory framework by classifying the substance according to its intended use (the label statement); the explicit or implied claim as the result of the label statement; how the use then defines the regulatory category; and ultimately, the safety standard (Figure 2).
Definitions According to the Food Additive Amendment
When the Food Additive Amendment (FAA) became law in 1958, the first task was to define the categories to be regulated in various parts of §201 (i.e., drugs, foods, food ingredients), and in 1994, with the passage of the Dietary Supplement Health and Education Act, the role of dietary supplements was defined in §201(ff) as distinct from drugs.
The Definition of a Drug
The definition of a drug is a functionality definition, and, for our purposes, in §201(g)(1) of the FAA: “The term ‘drug’ means…(B) articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals; and (C) articles (other than food) intended to affect the structure or any function of the body of man or other animals.” Therefore, on the label (or in the information media including, but not limited to broadcast, print, or social media), any mention of the five words “diagnosis, cure, mitigation, treatment, or prevention” plus identification of a disease makes the substance a drug. The carve-out of “other than food” was necessary because foods can cure, prevent, or mitigate diseases as described in the first paragraph. There is also a provision in this definition for “a food or dietary supplement for which a claim … is made … is not a drug solely because the label or the labeling contains such a claim,” although the claim could not cite the five words + disease name. It is important to note that a substance that has once been the subject of an Investigational New Drug (IND) application or has undergone clinical studies that have been published is precluded from use as a dietary supplement or addition to food.
The Definition of a Food
Food, in §201(f) of the FFA, is defined largely in terms of its functionality as well and is divided into three sections: “(1) articles used for food or drink for man or other animals, (2) chewing gum, and (3) articles used for components of any such article.”
The first part of the definition, “articles used for food or drink for man or other animals,” refers to processed food, such as angel food cake, hot dogs, or processed cheese and unprocessed food such as meat, eggs, and produce. A list of food categories into which the candidate food must fit, is provided in 21 CFR 170.3(n)(1-43). This list of food categories dates from the early 1970s and is quite limited in scope; however, a considerably more comprehensive list of food categories and their consumption data for the purpose of determining exposure should be available from your consultant. A simple list of food categories and serving sizes will grossly underestimate exposure, which may potentially result in harm to the consumer. All foods must be clearly identified as such and, in addition, a finished food will have a label listing the ingredients and a “Nutrition Facts” panel.
Chewing gum, the second part of the definition of food, was the basis of an argument as to whether it was swallowed or expectorated; the “swallower” contingent won out, so chewing gum was included in the food definition. Ironically, breath mints, although having much the same function as chewing gum, were not included in the food definition because they are considered a cosmetic.
The third part of the food definition—“articles used as components of any such article”—refers to any ingredient going into the finished food mentioned above, i.e., “articles used for food or drink… .” In the statute (§201(s)), these ingredients (“articles”) are referred to as becoming a component or otherwise affecting the characteristics of a finished food and are listed on the ingredient label.
These ingredients to be added to food must have a functionality (or technical effect) consistent with the list provided in 21 CFR 170.3(o)(1–32). Again, this list is fairly limited, and an experienced consultant can help provide better definition for the technical effect because the technology of food ingredients has evolved over the past 50 years.
The Definition of a Dietary Supplement
So what about the definition of a dietary supplement? According to the FFA (§201(ff)), a dietary supplement is both object specific and functionality based. It is object specific as it names vitamins, minerals, herbs or other botanicals or a concentrate, metabolite, extract thereof, etc., as dietary supplements, with a specific exclusion for tobacco. It is also a function-based definition because the phrase “a dietary substance for use by man to supplement the diet by increasing the total dietary intake” is included, meaning that the candidate substance must have been present in the diet historically and that the supplement cannot be represented as a conventional food or as the sole item of a meal, to discourage a diet consisting solely of supplements. Also, a dietary supplement label must include a “Supplement Facts” panel, required by the Dietary Supplement Health and Education Act of 1994.
So where, if at all, does a requirement for “natural” fit into the above definitions? It fits in only one: dietary supplements. That is, with a couple of exceptions, to be a dietary supplement ingredient, a supplement (or principal ingredient thereof) must be (or have been) present in the human diet historically because a dietary supplement is used to “increase the total dietary intake” and therefore, if not having been present in (unprocessed) food, it cannot be a dietary supplement ingredient. Also, the substance must “remain” a natural; that is, a natural substance cannot be chemically altered (although a few exceptions are permitted).
Safety as a Relative Concept
• The Safety Standard for Drugs. The standard for safety is relative to the regulated product. For example, the safety of a drug is given in §201(p)(1) “… as safe and effective for use under the conditions prescribed, recommended or suggested in the labeling thereof …” Therefore, a drug is safe if it is being used safely … a benefit versus risk standard. That is, the benefit derived from the use of the drug should not exceed the risk of its use. An example would be the use of injectable hydrocortisone for mild muscular pain. The risk associated with such a powerful steroid would include elevated blood glucose, suppression of the immune system, stimulation of gastric secretions and other undesirable effects, especially with chronic administration. Here the risks associated with use are greater than the benefit. Interestingly, although a drug is approved by the FDA for one or more specific uses, the prescribing physician is given considerable latitude and may prescribe an “off-label use,” i.e., a use that has not been approved by the FDA.
• The Safety Standard for Foods (and Food Ingredients). For foods, in FAA §201(u): “the term ‘safe’ [for food] … has reference to the health of man or animal.” This definition is expanded upon in the regulation (21 CFR §170.3(i)): “Safe or safety means that there is a reasonable certainty in the minds of competent scientists that the substance is not harmful under the intended conditions of use.” There are three key parts in this CFR elaboration. (1) The rule talks about “competent scientists,” which is further elaborated upon at the FDA website Draft Guidance for Industry: Best Practices for Convening a GRAS (Generally Recognized as Safe) Panel; that is, a person must be competent and not biased. (2) “Reasonable certainty” is the safety standard because as the regulation (21 CFR §170.3(i)) notes: “It is impossible in the present state of scientific knowledge to establish with complete certainty the absolute harmlessness of the use of any substance.” (3) “Intended conditions of use” embodies the concept that it is the use of the substance that is approved (i.e., for a particular use, for use in one or more particular food categories, and at a particular level of use); that is, the approval via a GRAS or food additive petition, is the use of the substance, not the substance itself for any food category, purpose, or at any concentration. However, like drugs with a provision for “off-label use,” food ingredients may be used for nonapproved purposes, if the use is “safe and suitable.”
Unlike drugs, there is no benefit versus risk for foods; all foods must be safe for the average person, although there are certain exceptions such as for allergens (e.g., soy, shellfish, eggs), which require warning labels under the Food Allergen Labeling and Consumer Protection Act of 2004 and the occasional substance for which a tolerance has been established, such as for sugar alcohols, polydextrose, or gluten.
While food is defined in §201(f) for more or less anything that is treated as food, the safety standard for unprocessed food (e.g., meat, eggs, produce) is not identified in the FAA until §402 (a)(1) wherein it is stated: “A food shall be deemed to be adulterated: (a)(1) If it bears or contains any poisonous or deleterious substance which may render it injurious to health; but in case the substance is not an added substance such food shall not be considered adulterated under this clause if the quantity of such substance in such food does not ordinarily render it injurious to health.” And, “or (a)(3) if it consists in whole or in part of any filthy, putrid, or decomposed substance, or if it is otherwise unfit for food… .”
Therefore, a food (processed or not) is adulterated if it bears or contains a substance that makes it injurious to health, unless the substance is not an added substance; that is, a food may contain a non-added (i.e., naturally present), deleterious (i.e., poisonous, toxic, or carcinogenic) substance so long as the conventional use of the food does not result in injury (otherwise, it would be unlikely to be called a “food”). In some instances, FDA has placed upper limits (called tolerances or action levels), wherein a food containing a toxic substance that is not an added substance and might have been formed or incorporated into the food during growth and normal metabolic processes essential to the normal development of the organism but not as the result of human actions. Therefore, in the absence of an added toxin, the food is considered safe, so long as it is not rotten, decomposed, or unfit for consumption as food.
In the broad view, foods themselves are presumed to be safe (subject to §402, above). An example would be a pea or carrot to be added to canned beef stew as an already GRAS ingredient for addition to the stew and not requiring a separate GRAS determination of a food additive petition. In regulatory parlance, there is a presumption of safety of food.
• The Safety Standard for Dietary Supplements.
The safety standard for a dietary supplement in FAA §413(a) is that “… the dietary supplement will reasonably be expected to be safe…” based on the opinion of the submitter, i.e., there is no requirement for demonstration of scientific expertise (i.e., analogous to “competent scientists” for a GRAS determination) of the submitter when submitting a New Dietary Ingredient (NDI) Notification to FDA. The safety standard of reasonable expectation, although worded differently than the food ingredient standard of reasonable certainty is seen by FDA as a distinction without a difference; that is, a substance is either safe or it is not. The author agrees with the FDA binary standard of safe versus not safe, but believes the FDA fails to recognize the distinction between reasonable expectation and reasonable certainty as being analogous to the evidentiary standards of a “preponderance of evidence” versus “clear and convincing evidence.” That is, if the latter (clear and convincing) is equivalent to the food standard of reasonable certainty, then the dietary supplement standard should be equivalent to “preponderance of evidence.” Therefore, the evidence of safety for reasonable expectation might not be as clear-cut and compelling as the reasonable certainty standard, but the reasonable expectation standard points persuasively to a positive outcome from exposure to the supplement, and there is no evidence contraindicating a safe exposure. The reasonable expectation level of evidence is greater than a simple lack of reports of adverse effects, but an affirmative finding of safety whose supporting body of evidence is less than comprehensive as it might be for food (i.e., clear and convincing evidence).
An important distinction between the arbiters of safety for a food ingredient and a dietary supplement is the expertise required; that is, the statutory requirement for a GRAS conclusion is “…experts qualified by scientific training and experience…,” but there is no equivalent standard for determining what constitutes reasonable expectation of safety for a dietary supplement. FDA has plugged what would seem to be this substantive loophole, by requiring the NDI Notification to undergo review by the agency prior to filing—essentially a premarket approval process, something the framers of the Act had sought to avoid.
Classification—Intended Use Drives the Category, Which Determines the Safety Standard
It has been said that the FDA, as an agency, is primarily concerned with labeling and safety. As mentioned above, the intended use of the substance is stated on the label (e.g., therapeutic effect, nutritional effect). This intended use determines the regulatory category (e.g., drug, food, or supplement), which in turn, determines the safety standard (see Figure 2).
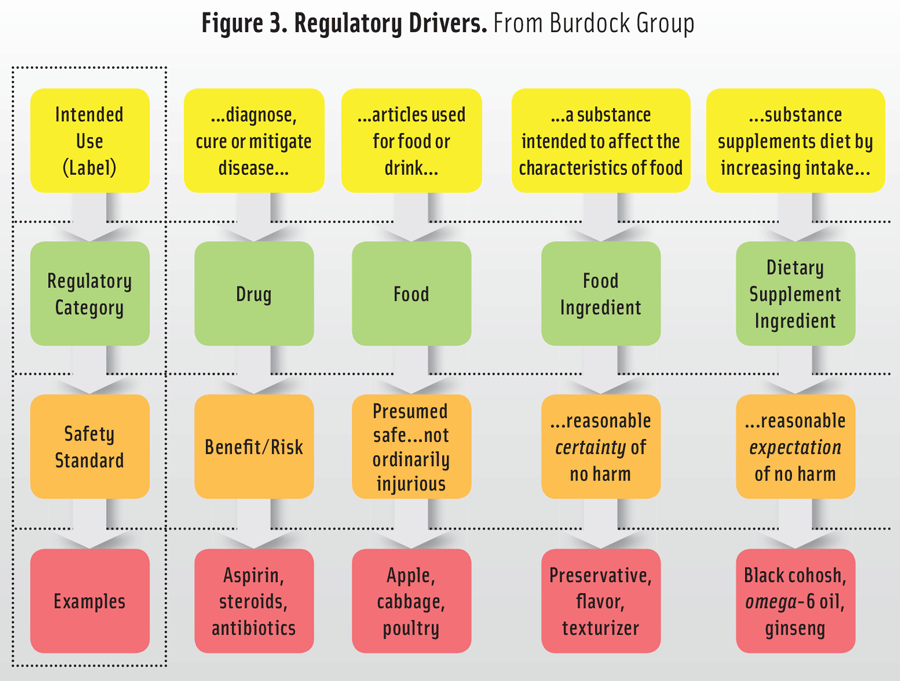
Figure 3. Regulatory Drivers. From Burdock Group
Therefore, when considering a read-across-like illustration of the relationship of intended use, consequent regulatory category, and the appropriate safety standard, the comparison appears as shown in Figure 3. For the top row, there is a summary of the statutory (or regulatory) language, e.g., diagnose, cure, or mitigate disease for a drug; articles used for food or drink are foods; a substance intended to affect the characteristics of food are food ingredients; if a substance supplements the diet, it is a dietary supplement. Each of the down arrows identifies the result of the information provided in the preceding row—the intended use (the label) determines the regulatory category, which controls the safety standard (e.g., benefit/risk, a presumption of safety and not ordinarily injurious, reasonable certainty of no harm, or a reasonable expectation of no harm).
While there are certainly exceptions, this grid with its pathways can direct the efforts of the regulatory and marketing experts within a company. For example, an added ingredient whose functionality is meant to alter the texture of a processed food (i.e., “… a substance intended to affect the characteristics of the food…”) to which it is added is a food ingredient. The matrix indicates the safety standard is a “… reasonable certainty of no harm… .” Say, for example, a manufacturer synthesizes a substance analogous to lycopene (an antioxidant)—which pathway should be taken? While an antioxidant would be a candidate as a food ingredient or a dietary supplement, the fact that the candidate substance is synthesized, means it cannot be assumed to be a “substance [that] supplements [the] diet by increasing the intake.” In other words, the substance was not in the diet as a normally present constituent of food and cannot be a dietary supplement. The pathway must be as a food ingredient.
Any R&D department, entrepreneur, or just someone with a good idea to improve the lives of consumers with a new product needs to know the regulatory environment because what is stated on the label will determine the regulatory thresholds for that particular product and the cost and depth of testing required to meet that regulatory threshold. Ignorance, willful or not, can doom the best of products.
George Burdock, PhD, is president, Burdock Group Consultants, an Orlando, Fla.–based safety and regulatory consulting firm (gburdock@burdockgroup.com).
Hero Image: © CharlieAJA/iStock/Thinkstock
Authors
-
George Burdock