
Nutrigenomics : An Emerging Scientific Discipline
The identification and understanding of individual and population differences and similarities in gene expression in response to diet can lead to food products customized for an individual’s nutritional needs.
Effects of the Human Genome Project are surfacing in anticipated and unanticipated areas. One of the key discoveries from the project is the existence of individual differences in gene sequences that result in differential response to environmental factors, such as diet. Those genetic differences, single nucleotide polymorphisms (SNPs, pronounced snips), are the key genetic enabler of the emerging scientific discipline called nutrigenomics or nutritional genomics. In the broad arena of health and wellness, one of the major impacts on the food industry, throughout the value chain from agricultural crops through consumer products, is this emerging understanding of human nutrition at the molecular level of gene expression.
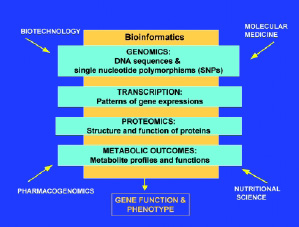 The science of nutrigenomics is the study of how naturally occurring chemicals in foods alter molecular expression of genetic information in each individual. In addition to bioinformatics, three scientific methodological approaches underpin nutrigenomics. Those methodological approaches are based on nutrition, molecular biology, and genomics. Integration of these disciplines is leading to identification and understanding of individual and population differences and similarities in gene expression, or phenotype, in response to diet (Fig. 1). The implications for the food industry range from market opportunities for customized or nutrient-enhanced crops through to products customized for individual nutritional needs.
The science of nutrigenomics is the study of how naturally occurring chemicals in foods alter molecular expression of genetic information in each individual. In addition to bioinformatics, three scientific methodological approaches underpin nutrigenomics. Those methodological approaches are based on nutrition, molecular biology, and genomics. Integration of these disciplines is leading to identification and understanding of individual and population differences and similarities in gene expression, or phenotype, in response to diet (Fig. 1). The implications for the food industry range from market opportunities for customized or nutrient-enhanced crops through to products customized for individual nutritional needs.
The Science
Scientists have long suspected that genetic differences among individuals were responsible for variations in response to environment, particularly dietb�0;14;which is arguably one of the major, constant environmental factors to which our genes are exposed throughout life. Those variations allowed some individuals to consume diets that seemed destined to lead to chronic disease but did not, while other individuals consuming similar diets evidenced expected or even more aggressive chronic disease patterns.
When a gene is activated, or expressed, a protein is produced. Through molecular biology and the tools of genomics, scientists have identified genes responsible for production of nutritionally important proteins such as digestive enzymes, transport molecules responsible for ferrying nutrients and cofactors to their site of use, and numerous other molecules responsible for metabolism and utilization of our dietary components, including macronutrients, vitamins, minerals, and phytochemicals.
Gene expression patterns produce a phenotype, which represents the physical characteristics or observable traits of an organism, e.g., hair color, weight, or the presence or absence of a disease. Phenotypic traits are not necessarily produced by genes alone. Phenotypic expression is influenced by nutrition. For example, diets alter cholesterol levels and types (LDL, HDL, and their ratios), homocysteine levels, and obesity (nutritional component), and these responses differ among individuals (genetic component).
At the molecular level, variations in one DNA building block result in variations in gene structure. That variation, or SNP, can lead to variations in the protein structure after the gene or its variant is expressed. Some structural variations in a protein may have an impact on its function, while some may not. Multiple genes may contain one or more SNPs, in distinctive patterns, associated with phenotypic patterns of nutritionally related health states, such as homocystenemia, high (or low) cholesterol, or variations in HDL and LDL cholesterol and their subunits. Patterns of multiple SNPs are called haplotypes.
The key element that distinguishes nutrigenomics from nutrition research is that the observable response to diet, or phenotype, is analyzed or compared in different individuals (or genotypes). Classical nutrition research essentially treats everyone as genetically identical, even while realizing that some individuals require more or less of specific nutrients. Molecular biology and biochemical approaches often assume the same: that an enzyme or flux through a metabolic or signal transduction pathway is the same in each individual. Similarly, the key element that distinguishes nutrigenomics from both molecular and genomics research is that nutrigenomics analyzes genetic expression in response to variations in diet (Fig. 2 on p. 62).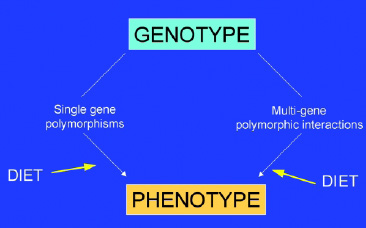
--- PAGE BREAK ---
Molecular and genomic research often assumes that the environment does not influence genetic expression. Nutrigenomics combines these concepts. An individual enzyme, pathway, or collection of pathways will be unique to groups or each individual, depending on the variation (defined by SNPs, haplotypes, and other polymorphisms) inherited. The expression or activity of these variant forms of normal genes differs depending on the amount and type of food ingested and the interactions between the food and the specific genotype.
Understanding these interactions has significant implications: turning genes on or off or changing the abundance of certain genes in response to different dietary chemicals may affect the balance between health and illness. Causes of chronic diseases, such as cardiovascular disease, cancer, or cognitive decline in aging, are not well understood because they are multi-factorial in nature. While we have clues to some dietary and other environmental or lifestyle factors that appear to contribute to occurrence of those and other chronic diseases, effects are not consistent among individuals. Thus, clear causeb�0;13;effect relationships are still emerging.
Occurrences of chronic diseases are b�0;1C;encodedb�0;1D; by a combination of factors, all acting on the body over time to create the disease phenotype of variable severity. These factors may include a number of genes, common genetic variants (i.e., SNPs, haplotypes), environmental factors, risk-conferring behaviors, and socioeconomic status. The genetic factors contributing to complex disease are difficult to identify because they typically exert small effects over long periods of time. Moreover, other unrelated genes and environmental factors, such as diet and lifestyle, can modify the magnitude of their effect.
Growth in Knowledge
Similar to the proverbial b�0;1C;blindfolded description of an elephant,b�0;1D; significant contributions from genomics, molecular biology, and nutritional disciplines have been made toward understanding the different components of complex, chronic disease phenotypes. However, a comprehensive, integrated picture still eludes us. This is specifically true for the effect of food on health and disease process. Epidemiological studies repeatedly have demonstrated associations between diet and cardiovascular disease, cancer, and other chronic diseases. However, the specific causeb�0;13;effect linkages between nutrient type and amount and health or disease phenotype are only beginning to emerge. The promise of nutrigenomics is that scientific research can deliver scientific evidence of health benefits of specific nutrients and foods for specific individuals or groups. Consumers are beginning to expect such assurances. The potential of nutrigenomics for the food industry is to provide good-tasting products formulated to the scientific targets. Consumers have and will continue to expect sensory satisfaction from foodsb�0;14;even foods for health.
Since health or disease processes are the common meeting point for genomic, molecular, and nutrition research, the remainder of this article will discuss nutrigenomics in light of the advances from these three disciplines in understanding disease, health, and wellness.
Genomic Analyses
The analogy of pharmacogenomics to nutrigenomics is readily evident. The goals of these areas are similar: customization of therapy, prevention and management of disease, and market segmentation based on personalized criteria. Through analysis of gene expression, SNPs, haplotypes, and biochemical and physiological results, scientists have verified individual and group differential responses to diet. This section will review selected examples of monogenic and complex, or polygenic, diseases, geneb�0;13;diet interaction, and implications for the food industry.
Genomic mapping and cloning strategies have revolutionized the process of localizing and identifying genes involved in monogenic disease and spurred the use of similar strategies for analyzing genes involved in complex diseases and phenotypes (Lander, 1996).
--- PAGE BREAK ---
To date, almost 1,000 human disease genes have been identified and partially characterizedb�0;14;97% of these genes are now known to cause monogenic diseases (Jimenez-Sanchez, 2001). Examples of monogenic diseases are sickle cell anemia, produced by a single amino acid change in hemoglobin that reduces hemoglobinb�0;19;s affinity for oxygen, and phenylketonuria, caused by a mutation in phenylalanine hydroxylase that leads to buildup of toxic levels of phenylalanine. However, the molecular basis of chronic disease is not fully understood, in spite of the more than 600 association studies published to date (Hirschhorn et al., 2002), the b�0;1C;failureb�0;1D; to identify single genes responsible for chronic diseases led to the common variant/common disease (CV/CD) hypothesis (Lander, 1996; Collins et al., 1997), which is largely responsible for the current genome-centric approach to the study of chronic diseases (Hirschhorn et al., 2002; Cargill and Daley, 2002; Taylor et al., 2001).
This hypothesis, in its simplest form, is that combinations of naturally occurring gene variants (i.e., alleles of unlinked genes) rather than mutations produce any given chronic disease. The link to nutrigenomics is that some of these naturally occurring gene variants will alter metabolism of nutrients, which in turn will alter the regulation genes involved in maintaining health or promoting disease.
Methods similar to those that identify single-gene defects have identified several minor, or low-penetrance, genes involved in chronic disease (Hirschhorn et al., 2002). An example of such a gene variant is the Pro12Ala polymorphism in the peroxisome proliferator activated receptor (PPAR-γ) associated with type 2 diabetes (Altshuler et al., 2000). However, numerous other genetic studies designed to identify chromosomal regions encoding genes involved in complex diseases have yielded inconsistent results. Indeed, in 600 association studies mentioned above, only six geneb�0;13;disease associations can be consistently replicated. The common study design errors in these studies include small sample size, poorly matched control groups, population stratification, overinterpretation of data, and others (Cardon and Bell, 2001; Lander and Kruglyak, 1995; Tabor et al., 2002; Risch, 1997). These methods and approaches are being improved to eliminate such errors and to reliably identify genes associated with complex phenotypes (Collins-Schramm et al., 2002; Parra et al., 1998; McKeigue et al., 2000; Deng et al., 2001; Pfaff et al., 2001; Reich and Goldstein, 2001).
Noticeably missing from discussions of the limitations of these genetic mapping techniques and gene association studies are the effects of environmental variables such as diet. Patterson et al. (1991) observed the importance of environment on expression of phenotypic traits in F2 and F3 generation tomato plants grown in Davis or Gilroy, Calif., vs plants grown near Rehovot, Israel. Only four of 29 quantitative trait loci (QTL) were found in all three sites, and 10 QTL were found in only two sites. QTL identify multiple regions within all chromosomes that collectively contribute to a complex phenotype (Risch et al., 1993). Since individual QTL encode many genes, identifying the causative genes within the QTL is challenging (Tabor et al., 2002). So far, the QTL mapping that accounts for differences in diet have not been done, largely because controlling for diet in large-population association studies is often not possible.
However, several recent association studies have included dietary variables in studies testing whether a single gene variant is associated with a complex phenotype:
-
Hypertension. A variant (designated AA) of the angiotensinogen (ANG) gene is linked with circulating ANG protein, which in turn, is associated with increased blood pressure. The Dietary Approaches to Stop Hypertension (DASH) diet positively affects individuals with the AA genotype, but the same diet was less effective in reducing blood pressure in individuals with a GG genotype. A large percentage (~60%) of African- Americans have the AA variant, with the remainder heterozygotic (AG) at this position (Svetkey et al., 2001).
- Cardiovascular Health. Apo-A1 plays a central role in lipid metabolism and coronary heart disease. A G to A transition in the promoter of APOA1 gene is associated with increased HDL-cholesterol concentrations, but the results across studies are not consistent. Ordovas et al. (2002) found that the A allele (or variant) was associated with decreased serum HDL levels. The genetic effect was reversed, however, in women who ate more polyunsaturated fatty acids (PUFA) relative to saturated fats (SF) and monounsaturated fats (MUFA). In men, this type-of-fat effect was significant when alcohol consumption and tobacco smoking were considered in the analyses. If confirmed by other studies, the APOA1 gene shows a classical geneb�0;13;environment interaction. Such interactions may help explain why candidate gene studies show inconsistencies. Food intake therefore may alter susceptibility to diseases mediated by increased HDL-cholesterol levels.
--- PAGE BREAK ---
-
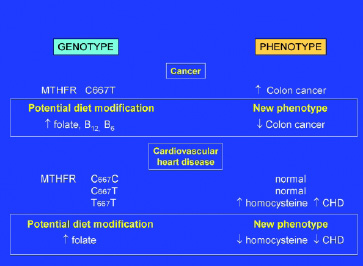
Cancer. Methylenetetrahydrofolate reductase (MTHFR) is a key gene in one-carbon metabolism and indirectly in all methylation reactions. Several laboratories have noted that the C667T polymorphism (ala to val), which reduces enzymatic activity, is inversely associated with occurrence of colorectal cancer (Slattery et al., 1999; Chen et al., 1999; Ulrich et al., 1999). Dietary recalls were used to assess intake of folate, vitamin B-12, vitamin B-6, or methionine (and in one study, alcohol) in individuals with the CC or TT phenotypes. Low intakes of these vitamins were associated with increased risk for cancer among those with the MTHFR TT genotype. MTHFR variants are also implicated in cardiovascular disease (Fig. 3 on p. 64).
Approximately 50 genetic diseases in humans are caused by defective enzymes (Ames et al., 2002). A subset of these enzymes are altered by naturally occurring SNPs which increase the Michaelis constant, Km, of coenzyme for enzyme. Km is a biochemical measure of the affinity of coenzyme (or substrate) for enzymeb�0;14;an increased Km results in decreased affinity. In certain cases, increasing the coenzyme concentration may ameliorate the decreased enzymatic activity. The medical applications for such cases would be that if genetic tests were available for the variant gene and if that variant was shown to be the only cause of a disease process, a physician or nutritional expert could recommend increasing or decreasing intake of a specific vitamin or food.
For example, increased dietary intake of nicotinic acid or nicotinamide might increase NADPH coenzyme concentrations enough to alter the equilibrium of GPDH b�0;6;�0;14; GPDHb�0;13; NADPH (Table 1). The same approach will not work for ALDH because the NAD substrate concentration could not be increased enough to overcome the increased Km caused by the substitution of lysine for glutamic acid at position 487.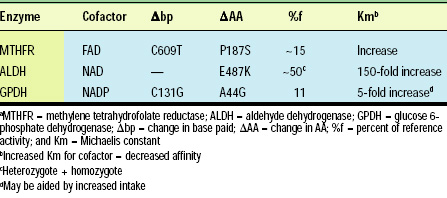
Elson-Schwab and Ames (2003) have established a Web site (www.kmmutants.org) that summarizes nutritional information for a large number of coenzyme-containing enzymes.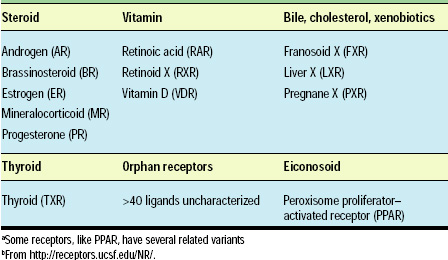
Molecular Analyses
Molecular studies in cell culture systems also have contributed details of signal transduction and biochemical pathways, nuclear receptors and transcription factors, and cell typeb�0;13;specific responses that contribute to, or are altered in many chronic diseases. For example, at least 47 proteins, including membrane receptors and transporters, kinases, phosphotases, and transcription factors, are involved in insulin signaling (www.grt.kyushu-u.ac.jp/spad/pathway/insulin.html or www.biocarta.com/pathfiles/h_insulinPathway.asp).
Since insulin affects many disparate pathways and physiological functions, the complexity is large and difficult to understand. Similar to the old biochemical pathway charts, these pathways are detailed as if the components are invariant. However, we now understand that the components vary. The Human Genome Project has demonstrated that variations in genes exist, and biochemical analyses have shown that such variations will alter enzymatic activities. As one example, steroid 5 -reductase (designated SRD5A2), a key enzyme in androgen metabolism in the prostate, has 13 naturally occurring variants in the human population. Nine of these variants reduce SRD5A2 activity by 20% or more, and three increase activity by more than 15% (Makridakis et al., 2000). Since SRD5A produces dihydrotestosterone (DHT) and DHT regulates genes in the prostate, variants in steroid 5 -reductase may affect incidence or severity of prostate cancer (Reichardt, 1999).
--- PAGE BREAK ---
A second limitation of molecular approaches is that diet or environment will also affect the expression (Cousins, 1999; De Caterina et al., 2001) and, in some cases the abundance, of the enzymes and proteins (Clarke and Abraham, 1992). Altering the concentrations of enzymes in the pathways will alter flux through pathways and ultimately the physiology of the organism.
Molecular studies have demonstrated that dietary chemicals or their metabolites bind directly to nuclear receptors (transcription factors) and alter gene expression (Table 2). For example, specific receptors exist for eiconosoids and vitamins (Lu et al., 2001; Dauncey et al., 2001; Aranda and Pascual, 2001; Chinetti et al., 2000; Chawla et al., 2001; Francis et al., 2002) (see http://receptors.ucsf.edu/NR/). In addition, some of these receptors have been shown to bind xenobiotics: the phytoestrogen genistein binds at a higher affinity to estrogen receptor ERβ compared to ERα (Kuiper et al., 1997). Since these receptors are expressed in many tissues throughout the body (Enmark et al., 1997), increasing genistein in the diet will have effects in multiple tissues.
Since each of these receptors has the potential to have variant forms, the effects of dietary chemicals on gene expression cannot be generalized from a single model to all humans. At present, the knowledge base of genetic variations is ahead of complete understanding of implications of those differences for nutritional biochemistry and health and wellness. While relationships are emerging, a vast amount of biology remains to be elucidated.
Nutritional Analyses
Environmental factors have long been known to contribute to development of chronic diseases, and these factors traditionally have been identified through epidemiological studies (Willett, 2002; Fleshner and Klotz, 1998; Maffeis, 1999). Such analyses, however, do not prove causality but rather identify potential environmental factors that are associated with incidence or severity of the disease. Separating the many variables in diet that might cause, mitigate, or promote disease is difficult. The key limitations for nutritional approaches are that the genotypes of individuals in any given study vary (see Genomic Analyses section), controlled feeding studies are expensive and logistically difficult to execute, and, as is well known in nutritional research, diet recalls are notoriously inaccurate.
In addition to the association studies that account for diet as a variable in the analyses, several laboratories have done classical nutritional crossover studies to begin dissecting dietary variables and their effects on specific metabolic responses and risk factors. That is, instead of dietary recalls, physiological analyses of metabolic parameters are determined after feeding different diets in the same individual:
Krauss and colleagues reported a series of studies (Krauss, 2001) showing that the low-density-lipoprotein (LDL) patterns are influenced by low-fat diets. They and others established that individuals with small, dense LDL particles (phenotype B) have an increased risk of coronary artery disease relative to those individuals exhibiting large, less dense LDL particles (phenotype A). Thirty-eight men exhibiting phenotype A LDL were switched from a diet containing 32% fat to one containing 10% fat. Twelve of these 38 exhibited phenotype B LDL after 10 days on the low-fat diet (Dreon et al., 1999), suggesting that for these 12, low-fat diets were not beneficial. These results should be interpreted with caution, since risk factors other than concentrations of LDL, triglycerides, HDL, and apolipoprotein A1 were not analyzed. That is, other risk factors, unassociated with serum lipid particles may be positively influenced by low-fat diets, and their contribution may be more important for long-term health. Nevertheless, these results suggest that a one-size-fits-all diet may not be appropriate.
Laboratory animal studies often reveal novel information that can be applied to human research. Rats and mice are particularly useful for such studies because they have relatively short generation times, their genetics have been intensively studied, and planned breeding can be done to test genetic hypotheses. Variables such as unique genotype can be controlled in studies with laboratory animals, and biological samples can be accessed more readily. On the other hand, most human molecular genetic studies are substantially more complex and difficult to execute. The human and mouse genome projects demonstrate the relatedness of the two species, and comparative genomic methods are being used to understand observed differences in physiology.
--- PAGE BREAK ---
Nutritionists have long used outbred or individual inbred strains of rats and mice for analyzing the effects of diet on health, which is equivalent to studying, respectively, populations or individuals. During the past 10 years, an experimental system based on comparative genomics was developed and tested that identifies genes involved in chronic diseases (Kaput et al., 1994; Park et al., 1997). The key to this strategy is to compare profiles of all genes (in one or more tissues) expressed differently between inbred strains of mice with different disease susceptibilities in response to control diet and to diets known to influence disease processes. A highly controlled feeding regimen ensures knowledge of the nutritional status at the time of sacrifice.
Inbred mouse strains represent the differences in disease susceptibilities observed between individual humans but allow for replication. Altering highly controlled environmental conditions, including diets, changes the regulation of genes that produce disease in susceptible mice but not in mice that are more resistant to the disease (Park et al., 1997). Once expression profiling or proteomic analyses identify diet-regulated genes, those genes are further analyzed by determining whether they are located within regions of chromosomes known to be involved in disease development. Several other laboratories have examined gene expression in model animals in response to diet, but the comparative approach is not routinely used.
The limitation of this approach had been that mice are not humans, and the results could not be extrapolated to humans. However, both the mouse (Waterston et al., 2002) and human (Lander et al., 2001; Venter et al., 2001) genomes are now sequenced, and it will be possible to use computational methods to predict responses in humans from the mouse data. Subsequent direct experimental verification will be necessary, but such experiments can be designed to be more inclusive and targeted.
Diet, Health, and Nutrigenomics
When asked, 75b�0;13;90% of consumers state that they make food choices with the intent of benefiting their or their familyb�0;19;s health. Health management and market segmentation through diet are possible, well established, and continue to grow.
Consumers seeking cholesterol management solutions have an array of foods available, including oatmeal, fat type and amount, carbohydrate type and amount, and stanols/sterols. Dairy products and soy for bone health, cancer, weight management, and cardiovascular health represent additional well-established and emerging health segments.
Dietary choices based on genetics are not new. Phenylketonuria and alcohol dehydrogenase deficiency are well-known conditions and can be avoided by avoiding consumption of phenylalanine and alcohol, respectively. Currently, food selection for health is based largely on generalized information, and, in some cases, more specific information derived from effects of diet on biomarkers such as bone density, cholesterol, serum triglycerides, or blood glucose, among others.
Epidemiological studies support the role of diet in health but have not revealed causeb�0;13;effect linkages that are emerging through the combination of the previously discussed scientific disciplines. Each of those disciplines contributes to unique, yet interrelated understanding of chronic disease and the role of diet in phenotypic expression of wellness or disease. The role of fatty acid intake and metabolism in depression (DHA/EPA); obesity, colon cancer, heart disease (PPARs); and partitioning of energy into adipose or muscle tissue (CLA) are but a few examples of geneb�0;13;diet interactions for which phenotype variability is being unraveled. Food clearly represents a nearly ideal channel through which to realize the benefits that nutrigenomics promises.
--- PAGE BREAK ---
Diagnostics
Diagnostics will play a critical and fundamental role in enabling nutrigenomics, both at the research level as well as at market introduction. For consumers, the initial introduction to practical application of nutrigenomics will be through diagnostics, initially genotyping, with identification of SNPs, followed by diagnostic monitoring of biomarkers to monitor effects of dietary changes. Presently, biomarker diagnostics such as cholesterol levels are more readily available and familiar to consumers. Genetic testing for the general population is becoming available, albeit the number of SNPs validated for dietary effects on phenotype is limited.
The Human Genome Project and subsequent SNP and haplotype (HAP) map projects are revealing that although all humans are 99.9% identical, the 0.1% difference produces significant differences. A subset of these differences is purely cosmetic (skin and hair color, size, etc). In seeking the SNPs and haplotype patterns that reveal dietary effects on phenotype, it is conceivable that scientists will discover other subsets of SNPs that may modify individual sensitivity to the otherwise safe chemicals, such that adverse reactions to some isolated generally recognized as safe (GRAS) chemical or combinations of chemicals may occur.
A few cautionary examples exist: for 46% of the end-stage renal patients, a Chinese herb (Aristolochia fangchi) used for appetite suppression reportedly contained nephrotoxins and urothelial carcinogens (www.cfsan.fda.gov/~dms/dsbotl3.html); and an L-tryptophan preparation contained contaminants causing eosinophiliab�0;13;myalgia syndrome in more than 1,500 individuals (Talalay, 2001). Some of these chemicals will induce damage regardless of the genotype of the individual. Other individuals may have and show susceptibility because of a single gene defect or variant in one key pathway. For cases where a single gene or limited number of genes is involved in the adverse reaction, genetic testing will be able to identify individuals susceptible to one or more chemicals.
However, it is also likely that certain chemicals will induce damage only in individuals having an unlucky combination of SNP (or other polymorphisms) in multiple genes. The b�0;1C;averageb�0;1D; dietary chemical interacts with membrane, cell, and serum transporters, may be metabolized to produce other bioactive compounds, or may alter regulation via specific or nonspecific interactions, e.g., selenium (Suzuki and Ogra, 2002). Proteins or enzymes that mediate any of these individual steps will likely have naturally occurring variations that contribute to the overall metabolic effect of an individual chemical. Hence, at least some adverse reactions will not be caused by a single gene defect or variant but by the combination of many gene products in different pathways involved in the metabolism of the chemical. In these cases, it will be much more difficult to identify all genes involved and the combination of SNPs responsible for the reaction.
Currently, several commercial entities are offering SNP analyses of b�0;1C;keyb�0;1D; genes involved in oxidant status, obesity or other complex phenotypes, even though no associated chronic (as opposed to monogenic) disease can be explained fully by these SNPs at the molecular level. Development of diagnostic testing to document genotype and SNPs and biomarkers to monitor dietary effects on gene expression, biochemistry, and physiology is clearly needed. Equally clear is the fact that this linkage of genotypeb�0;13;phenotypeb�0;13;biomarker diagnostic capability is beginning to occur and will be fundamental in the translation of nutrigenomics beyond the laboratory to the consumer market.
The most immediate application of gene testing may not be for a complex phenotype such as obesity or diabetes, but may be for b�0;1C;subphenotypesb�0;1D; such as insulin levels, glucose tolerance test result, or some defined and well-accepted b�0;1C;intermediateb�0;1D; biomarker of a disease. Genes that have the most influence on these subphenotypes may be more easily identified and therefore may be developed as diagnostics for predicting effects of specific food intake on specific subphenotypes.
--- PAGE BREAK ---
Two precautions are worth noting:
(1) each set of such genes defines only one subphenotype, and chronic diseases show complex sets of different subphenotypes (an example is the cluster of symptoms called Syndrome X whose symptoms have significant overlap with symptoms found in obesity, type 2 diabetes, and cardiovascular disease (Meigs, 2000); and (2) there are likely to be certain populations which have such different subphenotypes caused by different genetic contributions.
The promise of genetic testing for general nutrient intake and phenotype effects may not be met for some considerable time, since many of the association studies necessary to test the genes and dietary chemicals are costly and time consuming. As noted above, at present the knowledge base of genetic variations is ahead of complete understanding of implications of those differences for nutritional biochemistry and health and wellness. A vast amount of biology remains to be elucidated.
Epidemiological studies have associated serum levels of Creactive protein, cholesterol, and homocysteine, along with high serum levels of fibrinogen, and low serum albumin levels (Danesh et al., 1998), to risk of cardiovascular disease (Pearson et al., 2002; Grundy et al., 1999). In any given study, however, these risk factors may or may not be associated with overt disease. As noted above, different combinations of subphenotypes may produce the same overall disease pattern. Hence, the American Heart Association considers these markers as conditionally important in cardiovascular disease (CVD) and had not recommended routine measurements (Pearson et al., 2002; Grundy et al., 1999). Testing for level of C-reactive protein was recommended in late January 2003 for individuals with specific clinical conditions. LDL phenotype patterns A and B provide examples of how these other CVD-linked conditional biomarkers could be affected by diet and genotype or their interactions (Dreon et al., 1999). Until the regulation of the genes involved occurs and the effects of diet and environment are better characterized, it will not be possible to link SNP or other diagnostics to levels of these markers.
One has only to evaluate monthly scientific publications to see that a considerable scientific effort has been and is underway to unravel these associations. As the body of knowledge continues to develop, entrepreneurs and corporations with foresight will seize the early opportunities to begin creating and capturing the nutrigenomics value chain.
Regulatory, Legal and Ethical Considerations
Knowledge resulting from the scientific combination that underpins nutrigenomics leads to a potential change in theB borderline between medicine and foods, as defined by the Food and Drug Administration. The distinction between those current definitions will be challenged with growing evidence of nutrient effects on disease processes at the cellular level and a role for nutrients in disease prevention and management.
Modern pharmaceuticals evolved from thousands of years of traditional lore concerning the uses of plants and herbs as medicines. Research efforts over the past 100 years have led to the widespread adoption of Paul Ehrlich's Principles of (Chemo)therapy (Talalay, 2001):
- Drugs need not be of natural origin and could be developed by planned chemical synthesis.
- Systematic exploration of structure/function relationship distinguishing therapeutic activity from toxicity is needed.
- Maximization of ratio of dose required to cure disease to that producing toxicity (broad therapeutic index) is needed.
- The importance of developing animal models of diseases for quantitative measurements of both therapeutic potency and toxicity is needed.
--- PAGE BREAK ---
Highly sophisticated methods are now used to identify, characterize, and test potential drugs for effectiveness in humans to meet the criteria of these principles. However, the growing interest, acceptance, and use by the American public of dietary supplements, not to mention herbal medicines (Blendon et al., 2001), has outpaced the scientific, medical (Silverstein and Spiegel, 2001), and food industryb�0;19;s ability and capacity to carefully analyze the chemicals, their combined and independent activities, and their effectiveness and safety (Talalay, 2001; Matthews et al., 1999).
Although government regulation to ensure purity, truth in labeling, safety, and effectiveness began with the Pure Food and Drugs Act of 1906, with major revisions in 1938 (the Federal Food, Drug, and Cosmetic Act) and 1962 (Harris-Kefauver amendments), the Dietary Supplement Health and Education Act (DSHEA) of 1994 essentially terminated all government controls of safety and efficacy for dietary supplements (Talalay, 2001). DSHEA changed the regulatory climate, shifting to FDA the responsibility for proving a product unsafe, while relieving manufacturers of the responsibility to prove safety and efficacy.
It is beyond the scope of this article to explore the ramifications of that shift, but suffice it to say that the need for safety and efficacy data will only increase with the application of nutrigenomics approaches and methods. Since traditional nutrients and some phytonutrients will be considered as GRAS, the distinction between medicine and food will be further blurred. Nutrigenomics, by definition, will require clinical validation of effects, including safety, in the target market segment. Clearly there is an opportunity and a growing need for consideration of a regulatory framework that will accommodate emerging science as well as deliver consumer benefits and afford consumer protection.
The combination of emerging science of nutrigenomics and the weight of scientific evidence over the eight years since DSHEA was enacted strongly suggests that new regulatory frameworks are needed. While scientists suspected that food, like drugs, had cellular-level effects, the extent of that truth could not be supported with scientific evidence. Today, the proof is here and is growing. We are not suggesting that food be regulated as drugs. We are suggesting that thoughtful consideration be given to heretofore unexpected effects of nutrients and foods on health. Such consideration, if managed with foresight, ideally would support research to identify genes, adjunct diagnostic tests, and foods that would afford opportunity for optimal consumer health and wellness.
Translation to the Market
Marketing, selling, and delivering the benefits of nutrigenomics will require multiple disciplines, market channels, and points of consumer contact. Market segmentation for health and wellness based on dietary choices already is occurring according to dietary composition and health effects. Examples include cardiovascular health and oats, stanols/sterols, and fat amount and type.
At the consumer level, nutrigenomics will first be encountered as diagnostic testing for genetic patterns of SNPs, coupled with food products or supplements, and diagnostic monitoring of biomarkers that will track genetic response to diet. Consumer counseling will be essential to translate the meaning and recommended actions suggested by oneb�0;19;s genetic profile.
Successful incorporation of any food into an individualb�0;19;s diet will depend completely on whether the food fits an existing dietary pattern and has excellent sensory properties. In other words, like many other health trends in the food industry, nutrigenomics will thrive and deliver the benefits inherent in the concept only if the products deliver consumer benefits and satisfy consumer preferences.
Questions of ethics, privacy, compliance, insurance reimbursement, value creation and capture, and economic return, and, as noted above, the need for additional physiological and biochemical studies to identify and validate effects of dietary modification on phenotypic expression must be resolved.
The science from various disciplines that constitute nutrigenomics continues to emerge and is being integrated into useful information on which the food industry can act. Clearly, this is not the food industry as is has been in the past. Alliances, partnerships, or consortia among varied commercial partners will be essential to deliver on the scientific and commercial potential of nutrigenomics.
by Nancy Fogg-Johnson and Jim Kaput
Author Fogg-Johnson, a Professional Member of IFT, is Principal, Life Sciences Alliance, 796 Panorama Rd., Villanova, PA 19085. Author Kaput is Chief Scientific Officer and CEO, NutraGenomics, 7628 Garden Ln., Justice, IL 60458, and Senior Scientist, Center of Excellence in Nutritional Genomics, University of California at Davis. Send reprint requests to author Fogg-Johnson.
References
Altshuler, D., Hirschhorn, J.N., Klannemark, M., Lindgren, C.M., Vohl, M.C., Nemesh, J., Lane, C.R., Schaffner, S.F., Bolk, S., Brewer, C., Tuomi, T., Gaudet, D., Hudson, T.J., Daly, M., Groop, L., and Lander, E.S. 2000. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 26(1): 76-80.
Ames, B.N., Elson-Schwab, I., and Silver, E.A. 2002. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): Relevance to genetic disease and polymorphisms. Am. J. Clin. Nutr. 75: 616-658.
Aranda, A. and A. Pascual, 2001. Nuclear hormone receptors and gene expression. Physiol. Rev. 81: 1269-1304.
Blendon, R.J., DesRoches, C.M., Benson, J.M., Brodie, M., Altman, D.E. 2001. Americans’ views on the use and regulation of dietary supplements. Arch. Intern. Med. 161: 805-810.
Cardon, L.R. and Bell, J.I. 2001. Association study designs for complex diseases. Nat. Rev. Genet. 2(2): 91-99.
Cargill, M. and Daley, G.Q. 2002. Mining for SNPs: Putting the common variants–common disease hypothesis to the test. Pharmacogenomics 1(1): 27-37.
Chawla, A., Repa, J.J., Evans, R.M., Mangelsdorf, D.J. 2001. Nuclear receptors and lipid physiology: opening the X-files. Science 294: 1866-1870.
Chen, J., Giovannucci, E.L., and Hunter, D.J. 1999. MTHFR polymorphism, methyl-replete diets and the risk of colorectal carcinoma and adenoma among U.S. men and women: An example of gene-environment interactions in colorectal tumorigenesis. J. Nutr. 129(2S Suppl.): 560S- 564S.
Chinetti, G., Fruchart, J.C., and Staels, B. 2000. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 49: 497-505.
Clarke, S.D. and Abraham, S. 1992. Gene expression: Nutrient control of pre- and posttranscriptional events. FASEB J. 6: 3146-3152.
Collins, F.S., Guyer, M.S., and Charkravarti, A. 1997. Variations on a theme: Cataloging human DNA sequence variation. Science 278: 1580-1581.
Collins-Schramm, H.E., Phillips, C. M., Operario, D. J., Lee, J. S., Weber, J. L., Hanson, R. L., Knowler, W. C., Cooper, R., Li, H., Seldin, M. F. 2002. Ethnic-difference markers for use in mapping by admixture linkage disequilibrium. Am. J. Hum. Genet. 70: 737-750.
Cousins, R.J., 1999. Nutritional regulation of gene expression. Am. J. Med. 106(1A): 20S- 23S; discussion 50S-51S.
Danesh, J., Collins, R., Appleby, P., and Peto, R. 1998. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: Meta-analyses of prospective studies. J. Am. Med. Assn. 279: 1477-1482.
Dauncey, M.J., White, P., Burton, K.A., and Katsumata, M. 2001. Nutrition-hormone receptorgene interactions: Implications for development and disease. Proc. Nutr. Soc. 60(1): 63-72.
De Caterina, R., Madonna, R., Hassan, J., and Procopio, A.D. 2001. Nutrients and gene expression. World Rev. Nutr. Diet. 89: 23-52.
Deng, H.W., Chen, W.M., and Recker, R.R. 2001. Population admixture: Detection by Hardy- Weinberg test and its quantitative effects on linkage-disequilibrium methods for localizing genes underlying complex traits. Genetics 157: 885-897.
Dreon, D.M., Fernstrom, H.A., Williams, P.T., and Krauss, R.M. 1999. A very-low-fat diet is not associated with improved lipoprotein profiles in men with a predominance of large, low-density lipoproteins. Am. J. Clin. Nutr. 69: 411-418.
Elliott, T.S., Swartz, D.A., Paisley, E.A., Mangian, H.J., Visek, W.J., and Kaput, J. 1993. F1F0-ATPase subunit e gene isolated in a screen for diet regulated genes. Biochem. Biophys. Res. Commun. 190(1): 167-174.
Elson-Schwab, I., Poedjosoedarmo, K., and Ames, B.N. 2003. www.KmMutants.org.
Enmark, E., Pelto-Huikko, M., Grandien, K., Lagercrantz, S., Lagercrantz, J., Fried, G., Nordenskjold, M., and Gustafsson, J.A. 1997. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 82: 4258-4265.
Fleshner, N.E. and Klotz, L.H. 1998. Diet, androgens, oxidative stress and prostate cancer susceptibility. Cancer Metastasis Rev. 17(4): 325-330.
Francis, G.A., Fayard, E., Picard, F., and Auwerx, J. 2002. Nuclear Receptors and the Control of Metabolism. Ann. Rev. Physiol. Vol. 65.
Grundy, S.M., Pasternak, R., Greenland, P., Smith, S. Jr., and Fuster, V. 1999. AHA/ACC scientific statement: Assessment of cardiovascular risk by use of multiple-risk-factor assessment equations: A statement for healthcare professionals from the American Heart Association and the American College of Cardiology. J. Am. Coll. Cardiol. 34: 1348-1359.
Hirschhorn, J.N., Lohmueller, K., Byrne, E., and Hirschhorn, K. 2002. A comprehensive review of genetic association studies. Genet. Med. 4(2): 45-61.
Jimenez-Sanchez, G., Childs, B., and Valle, D. 2001. Human disease genes. Nature 409: 853-855.
Kaput, J., Swartz, D., Paisley, E., Mangian, H., Daniel, W.L., and Visek, W.J. 1994. Diet-disease interactions at the molecular level: An experimental paradigm. J. Nutr. 124(8 Suppl.): 1296S-1305S. Krauss, R.M., 2001. Dietary and genetic effects on LDL heterogeneity. World Rev. Nutr. Dietetics 12-22.
Kuiper, G.G., Carlsson, B., Grandien, K., Enmark, E., Haggblad, J., Nilsson, S., and Gustafsson, J.A. 1997. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138: 863-870.
Lander, E. and Kruglyak, L. 1995. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results [see comments]. Nat. Genet. 11(3): 241-247.
Lander, E.S. 1996. The new genomics: Global views of biology. Science 274: 536-539.
Lander, E.S. et al. (255 authors). 2001. Orphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J. Biol. Chem. 276: 37735-37738.
Maffeis, C., 1999. Childhood obesity: the genetic-environmental interface. Baillieres Best Pract. Res. Clin. Endocrinol. Metab. 13(1): 31-46.
Makridakis, N.M., di Salle, E., and Reichardt, J.K. 2000. Biochemical and pharmacogenetic dissection of human steroid 5 alpha-reductase type II. Pharmacogenetics 10: 407-413.
Matthews, H.B., Lucier, G.W., and Fisher, K.D. 1999. Medicinal herbs in the United States: Research needs. Environ. Health Perspect. 107: 773-778.
McKeigue, P.M., Carpenter, J.R., Parra, E.J., and Shriver, M.D. 2000. Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: Application to African- American populations. Ann. Hum. Genet. 64(Part 2): 171-186.
Meigs, J.B., 2000. Invited commentary: Insulin resistance syndrome? Syndrome X? Multiple metabolic syndrome? A syndrome at all? Factor analysis reveals patterns in the fabric of correlated metabolic risk factors. Am. J. Epidemiol. 152: 908-911; discussion 912.
Ordovas, J.M., Corella, D., Cupples, L.A., Demissie, S., Kelleher, A., Coltell, O., Wilson, P.W., Schaefer, E.J., and Tucker, K. 2002. Polyunsaturated fatty acids modulate the effects of the APOA1 G-A polymorphism on HDL-cholesterol concentrations in a sex-specific manner: The Framingham Study. Am. J. Clin. Nutr. 75(1): 38-46.
Park, E.I., Paisley, E.A., Mangian, H.J., Swartz, D.A., Wu, M.X., O’Morchoe, P.J., Behr, S.R., Visek, W.J., and Kaput, J. 1997. Lipid level and type alter stearoyl CoA desaturase mRNA abundance differently in mice with distinct susceptibilities to diet-influenced diseases. J. Nutr. 127: 566-573.
Parra, E.J., Marcini, A., Akey, J., Martinson, J., Batzer, M.A., Cooper, R., Forrester, T., Allison, D.B., Deka, R., Ferrell, R.E., and Shriver, M.D. 1998. Estimating African American admixture proportions by use of population-specific alleles. Am. J. Hum. Genet. 63: 1839-1851.
Patterson, A.H., Damon, S. Hewitt, J.D., Zamir, D., Rabinowitch, H.D., Lincoln, S.E., Lander, E.S., and Tanksley, S.D., 1991. Mendelian factors underlying quantitative traits in tomato: Comparison across species, generations, and environments. Genetics 127: 181-197.
Pearson, T.A., Blair, S.N., Daniels, S.R., Eckel, R.H., Fair, J.M., Fortmann, S.P., Franklin, B.A., Goldstein, L.B., Greenland, P., Grundy, S.M., Hong, Y., Miller, N.H., Lauer, R.M., Ockene, I.S., Sacco, R.L., Sallis, J.F. Jr., Smith, S.C. Jr., Stone, N.J., and Taubert, K.A. 2002. AHA guidelines for primary prevention of cardiovascular disease and stroke: 2002 Update: Consensus panel guide to comprehensive risk reduction for adult patients without coronary or other atherosclerotic vascular diseases. Am. Heart Assn. Science Advisory and Coordinating Comm. Circulation 106: 388-391.
Pfaff, C.L., Parra, E.J., Bonilla, C., Hiester, K., McKeigue, P.M., Kamboh, M.I., Hutchinson, R.G., Ferrell, R.E., Boerwinkle, E., and Shriver, M.D. 2001. Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am. J. Hum. Genet. 68: 198-207.
Reich, D.E. and Goldstein, D.B. 2001. Detecting association in a case-control study while correcting for population stratification. Genet. Epidemiol. 20(1): 4-16.
Reichardt, J.K., 1999. GEN GEN: The genomic genetic analysis of androgen-metabolic genes and prostate cancer as a paradigm for the dissection of complex phenotypes. Front. Biosci. 4: D596-600.
Risch, N., 1997. Evolving methods in genetic epidemiology. II. Genetic linkage from an epidemiologic perspective. Epidemiol. Rev. 19(1): 24-32.
Risch, N., Ghosh, S., and Todd, J.A. 1993. Statistical evaluation of multiple-locus linkage data in experimental species and its relevance to human studies: Application to nonobese diabetic (NOD) mouse and human insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 53: 702-14.
Silverstein, D.D. and Spiegel, A.D. 2001. Are physicians aware of the risks of alternative medicine? J. Community Health 26(3): 159-174.
Slattery, M.L., Potter, J.D., Samowitz, W., Schaffer, D., and Leppert, M. 1999. Methylenetetrahydrofolate reductase, diet, and risk of colon cancer. Cancer Epidemiol. Biomarkers Prev. 8: 513-518.
Suzuki, K.T. and Ogra, Y. 2002. Metabolic pathway for selenium in the body: Speciation by HPLC-ICP MS with enriched Se. Food Additives Contam. 19: 974-983.
Svetkey, L.P., Moore, T.J., Simons-Morton, D.G., Appel, L.J., Bray, G.A., Sacks, F.M., Ard, J.D., Mortensen, R.M., Mitchell, S.R., Conlin, P.R., and Kesari, M. 2001. Angiotensinogen genotype and blood pressure response in the Dietary Approaches to Stop Hypertension (DASH) study. J. Hypertens. 19: 1949-1956.
Tabor, H.K., Risch, N.J., and Myers, R.M. 2002. Opinion: Candidate-gene approaches for studying complex genetic traits: Practical considerations. Nat. Rev. Genet. 3: 391-397.
Talalay, P., 2001. The importance of using scientific principles in the development of medicinal agents from plants. Acad. Med. 76(3): 238-247.
Taylor, J.G., Choi, E.H., Foster, C.B., and Chanock, S.J. 2001. Using genetic variation to study human disease. Trends Mol. Med. 7: 507-512.
Ulrich, C.M., Kampman, E., Bigler, J., Schwartz, S.M., Chen, C., Bostick, R., Fosdick, L., Beresford, S.A., Yasui, Y., and Potter, J.D. 1999. Colorectal adenomas and the C677T MTHFR polymorphism: Evidence for gene-environment interaction? Cancer Epidemiol. Biomarkers Prev. 8: 659-668.
Venter, J.C. et al. (274 authors). 2001. The sequence of the human genome. Science 291: 1304-1351.
Waterston, R.H. et al. (222 authors). 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-62.
Willett, W.C., 2002. Balancing life-style and genomics research for disease prevention. Science 296: 695-698.
The Center of Excellence in Nutritional Genomics at the University of California at Davis is funded by National Institutes of Health grant No. NIH MD00222-01.